
Skeletal muscle atrophy,regeneration,and dysfunction in heart failure:Impact of exercise training
2023-09-26 09:00:32HarrisonGallagherPaulHendrickseMarceloPereiraScottBowen
Harrison Gallagher,Paul W.Hendrickse,Marcelo G.Pereira,T.Scott Bowen
School of Biomedical Sciences,Faculty of Biological Sciences,University of Leeds,Leeds LS2 9JT,UK
Abstract This review highlights some established and some more contemporary mechanisms responsible for heart failure(HF)-induced skeletal muscle wasting and weakness.We first describe the effects of HF on the relationship between protein synthesis and degradation rates,which determine muscle mass,the involvement of the satellite cells for continual muscle regeneration,and changes in myofiber calcium homeostasis linked to contractile dysfunction.We then highlight key mechanistic effects of both aerobic and resistance exercise training on skeletal muscle in HF and outline its application as a beneficial treatment.Overall,HF causes multiple impairments related to autophagy,anabolic-catabolic signaling,satellite cell proliferation,and calcium homeostasis,which together promote fiber atrophy,contractile dysfunction,and impaired regeneration.Although both wasting and weakness are partly rescued by aerobic and resistance exercise training in HF,the effects of satellite cell dynamics remain poorly explored.
Keywords: Calcium;Exercise training;Heart failure;Satellite cells;Skeletal muscle wasting
1.Introduction
Skeletal muscle is the most abundant tissue in the human body and is involved in various fundamental functions such as mobility (locomotion and posture),inspiratory function,thermoregulation,metabolism of macronutrients such as glucose,lipids,and amino acids,1and it has also been described as an endocrine organ.2Skeletal muscle tissue has a remarkable capacity to adapt to different stimuli(i.e.,plasticity),dramatically changing its mass and function according to each situation.While an increase in muscle mass (i.e.,hypertrophy)occurs in response to intense resistance exercise training(RET) or the presence of certain hormones,3loss of muscle mass and strength are often observed in specific scenarios,including physical inactivity,disuse,aging,and following chronic diseases such as cancer and heart failure(HF).4
Chronic disease-related muscle wasting at its most severe is often termed cachexia.Cachexia is defined as a complex,multifactorial metabolic syndrome underpinned by an underlying illness and associated with a significant reduction in body mass derived from muscle tissue loss with or without adipose tissue loss,and which cannot be reversed by conventional nutritional interventions.5Cachexia impairs quality of life in patients by reducing the effectiveness of treatments;indeed,evidence indicates that patients with cachexia exhibit shorter survival than non-cachectic patients.6,7In addition,cachexia also affects the main muscle of respiration,the diaphragm,8the wasting of which exacerbates symptoms of breathlessness and impairs ventilation,leading to life-threatening respiratory failure.9Nevertheless,it is important to recognize a large proportion of patients may not present with overt cachexia or wasting yet lose muscle strength due to intrinsic muscle dysfunction(i.e.,loss of function independent of mass).Accordingly,it is important to appreciate both aspects as key factors limiting quality of life in patients.10
Therefore,the recognition that chronic diseases inducing both muscle mass loss and dysfunction as a widespread condition affect millions of people has stimulated the search for treatments able to attenuate this and improve the quality of life of patients.While no effective pharmacological treatments are clearly established at present,exercise training has been proposed as a potential therapeutic approach due to its various effects on both the systemic and local muscle levels (i.e.,anti-inflammatory,immunological,11anti-atrophic,12,13and pro-oxidative metabolism14).In terms of chronic diseases,HF warrants important consideration because this condition continues to increase in prevalence,is one of the most common causes of hospitalizations15and global deaths,16and there is no cure.Of note,cardiac dysfunction in HF poorly correlates to symptoms and skeletal muscle dysfunction,which indicates that a more complex situation is at play.17,18
Therefore,in this brief review,we outline selected mechanisms underpinning limb and diaphragm muscle loss and weakness in HF,with a specific focus on fiber atrophy,regeneration,and contractile dysfunction.We then highlight the important benefits associated with exercise training for attenuating skeletal muscle impairments (as summarized in Fig.1).Unless otherwise noted,we focus mostly on studies of HF with reduced rather than preserved ejection fraction given that more evidence is available,which allows for more robust conclusions.
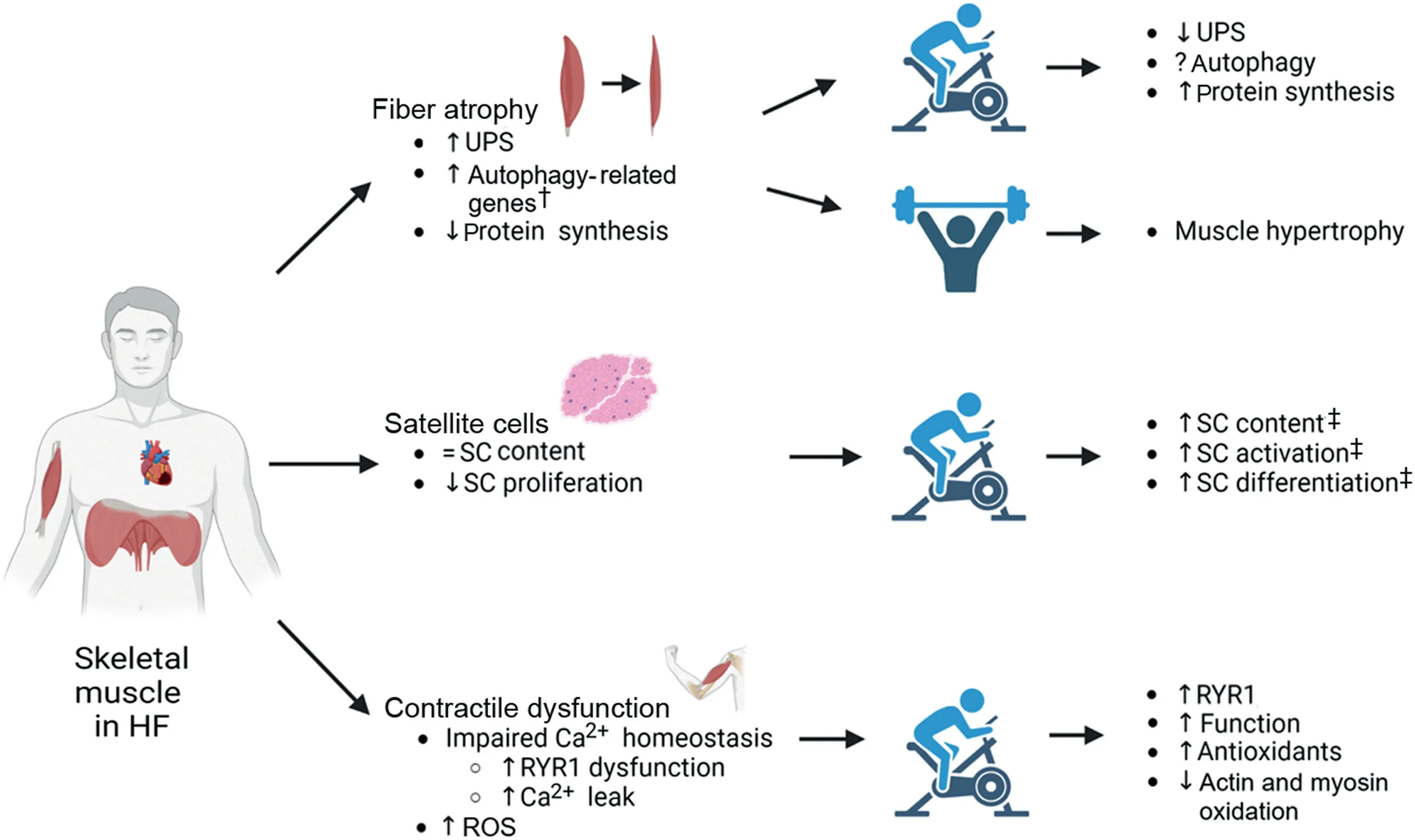
Fig.1.Summary of the primary effects of heart failure on skeletal muscle fiber atrophy,satellite cells,and contractile dysfunction,as well as the secondary impact following prolonged exercise training in patients and animal models.† means in animal models only;‡ means in healthy individuals.↑ means increase;↓ means decrease;=means no change;? means lacking information yet.HF=heart failure;ROS=reactive oxygen species;RT=resistance training;RYR1=ryanodine receptor 1;SC=satellite cell;UPS=ubiquitin proteasome system.
2.Effects of HF on fiber atrophy
The regulation of muscle mass and function reflects protein turnover(i.e.,the balance between protein synthesis and degradation).The 2 major proteolytic systems involved in muscle wasting are the ubiquitin proteasome system (UPS) and autophagy-lysosomal system (for a full review see Singh et al.19),although the calpain and caspase systems can also play important roles.20Whereas the UPS specifically degrades myofibrillar proteins,autophagy is responsible for the clearance of damaged cellular components via autophagosome formation.The UPS system is regulated by ubiquitin enzymes E1,E2,and E3,which respectively activate,carry,and bind ubiquitin to target proteins before degradation at the proteasome complex.The UPS is involved in a number of cachectic conditions,displaying high levels of E3-ligases as well as proteasome activity.Indeed,leg muscle samples(of the vastus lateralis (VL)) from HF patients display an increase in the protein content of E3-ligase muscle RING-finger protein-1(MuRF-1),21,22with concomitant increases in proteasome activity.23This finding was mirrored in a rat model of myocardial infarction (MI)-induced HF,where proteasome activity was higher in plantaris and soleus muscles.24However,given the limited access to patient samples,the role of autophagy in HF-induced cachexia remains less clear.It is known,however,that some autophagy-associated markers,such as the expression of microtubule-associated proteins 1A/1B light chain 3B and B-cell lymphoma-2 interacting protein 3,are upregulated in skeletal muscle during starvation periods,with forkhead box protein O3 recognized as the most important transcription factor controlling autophagy.25Despite evidence from experimental models of HF with preserved ejection fraction(HFpEF) and MI indicating autophagy may be dysregulated,26,27clinical patient data is limited.However,evidence indicates no difference in skeletal muscle mRNA expression or protein content of lysosomal proteolysis marker cathepsin L in patient tissue.22
As the underlying mechanisms of muscle wasting and HF progression remain poorly understood,an elegant study suggested the dysregulation of myokine expression from wasting muscles impairs HF severity.28The study observed that musclin expression is reduced in HF and the musclespecific disruption of musclin in mice contributes to the progression of HF,while elevated musclin levels improved cardiac function.28This is one of the first studies to suggest a link between skeletal-cardiac muscle cross talk in HF-induced muscle wasting that could suggest a promising therapeutic strategy.
Another mechanism that contributes to muscle wasting is an inability to activate pro-hypertrophic pathways,which is a condition known as anabolic resistance.In this sense,the key mediator of myofibrillar protein synthesis and muscle growth is the mechanistic target of rapamycin complex 1 (mTORC1)pathway.The activation of mTORC1 by upstream factorsinsulin-like growth factor-1(IGF-1) andprotein kinase B(Akt)phosphorylates downstream targetsribosomal protein S6 kinase beta-1,eukaryotic translation initiation factor4Ebinding protein 1,andeukaryotic initiation factor 4Eto activate protein translation.29It has been shown that HF patients with reduced or preserved ejection fraction alike displayed reduced skeletal muscle mRNA expression and protein content of IGF-1.21,30In line with this,phosphorylated Akt protein content is also lower in the skeletal muscle of HF patients,31which is perhaps indicative of impaired translational activity as it is in other conditions such as cancer.13One other key study in mice post-MI showed that muscle-specific overexpression of IGF-1 blocked atrophy via normalizing Akt phosphorylation in line with inhibiting E3-ligase expression and proteasome activity.32Interestingly,despite past evidence suggesting a poor link between cardiac dysfunction and skeletal muscle changes in HF,a study using dual X-ray absorptiometry showed left ventricular assist device recipients gained muscle mass within 6 months of surgery.33However,it remains unclear whether the increased muscle mass in HF patients was caused by increased blood flow or improvements in physical activity given the expected reduction in symptoms.Unfortunately,no muscle biopsies were taken to further investigate mechanistic signaling.Thus,it is important to consider 2 mechanistic angles in HF,both the hypertrophic and atrophic signaling nexus.
Alongside the mTORC1 pathway,the key upstream regulator of muscle protein balance is cell metabolism.Mitochondria regulate energy metabolism by integrating key cell signaling pathways related to oxidative stress and energy production.34Mitochondrial dynamics have been shown to regulate muscle mass in aging.It was observed that physical inactivity contributes to age-related decline in the activity of optic atrophy gene 1(OPA1),one of the genes regulating mitochondrial dynamics and biogenesis,which are associated with muscle atrophy.35It was also observed that a muscle-specific deletion of OPA1 alters mitochondrial morphology and function,leading to endoplasmic reticulum stress,which then induces a catabolic program via the unfolded protein response and forkhead box Os.35The role of mitochondria dynamics in the context of HF remains to be explored.However,evidence has shown that mRNA expression of OPA1 and peroxisome proliferator-activated receptor-gamma coactivator-1a were downregulated in female HF patients who also present impairedin situmitochondrial function.36Interestingly,these changes were not observed in male patients.Instead,male HF patients present functional impairments related to complex I oxidative phosphorylation,indicating an important divergence in phenotype between sexes.36
Accordingly,HF patients present a number of structural abnormalities in the mitochondria,including a reduction in size37and content36,38as well as fluid accumulation and membrane disruption.37In addition,HF patients with diabetes also present impairments in mitochondrial functionin situ,including reduced oxygen flux and coupling efficiency as well as a concomitant increase in reactive oxygen species (ROS)production.39Please refer to Lv et al.34for an in-depth review of mitochondrial dynamics in HF.
3.Effects of HF on satellite cells(SCs)and muscle regeneration
The changes in protein turnover leading to muscle hypertrophy or atrophy do not occur according to the simplistic balance between protein synthesis and degradation but may be affected by nuclear turnover.Hypertrophy can occur by accretion of new myonuclei by muscle stem cell or SC fusion,which in turn helps expand cytoplasmic volume,40while loss of myonuclei by cell apoptosis can lead to muscle atrophy.41Therefore,impairments to SCs may also contribute to reduced skeletal muscle mass in HF(Fig.1).
SCs are located between the sarcolemma and basal lamina of muscle fibers42and usually reside in a resting state known as quiescence,which is characterized by the expression of transcription factorpaired box 7(Pax7).43In response to exercise and/or muscle injury,Pax7is downregulated,and SCs enter an activated state.44In turn,SCs proliferate and differentiate under the control of a group of transcription factors termedmyogenic regulatory factors,with a proportion also returning to quiescence to repopulate the SC pool.45Differentiated cells fuse with one another to form new myofibers or with damaged myofibers to facilitate repair and myonuclear turnover.The understanding of the role of SCs in muscle has improved with the development of the Pax7-diptheria toxin A mouse model,which allows conditional ablation of SCs upon tamoxifen administration,46although this is yet to be tested in the context of HF.Despite proving critical for injury-induced skeletal muscle regeneration,47,48the role of SCs in muscle growth remains controversial.49,50Evidence suggests that SCs are required for optimal hypertrophy in aged muscle51and in response to chronic overload,52while impairments in SC function have been identified in patients and in various experimental models of muscle wasting.53,54
In the context of HF,only a handful of studies have explored the link between muscle atrophy and SCs.In a transgenic dilated cardiomyopathy mouse model,the plantar flexors were stimulatedin vivobefore their ability to regenerate following eccentric forced dorsiflexion contraction-induced damage was assessed.55The study showed that the plantar torque recovered by 95%within 2 weeks of injury in controls,but that it was attenuated in HF mice with the number of centrally located nuclei substantially elevated.55While these results suggest that HF impairs skeletal muscle regeneration after injury,the study concluded this was SC-independent given that the number of Pax7 positive cells was unchanged between groups.Clearly further experiments directly assessing SC function in HF will be needed to verify this assumption.Additional data confirmed that the SC number per 100 myofibers in the tibialis anterior muscle of obese HFpEF rats at baseline was not different when compared to lean controls.56In contrast,fiber size and SC abundance have been reported to be reduced in the gastrocnemius muscle post cardio toxininduced muscle injury following 7 days of treatment with angiotensin II(i.e.,a known peptide hormone playing a significant role in the development of cardiovascular disease).57Furthermore,to aid clinical translation and better explore the effects of HF on SC dynamics,the study confirmed that following MI in mice,the number of SCs in the gastrocnemius muscle was lower in comparison to that of the respective sham control group,while pharmacological blockade of the angiotensin Type I receptor prevented this.This further supports the viewpoint that angiotensin II could be an upstream trigger of myofiber atrophy in HF not only by regulating the transcription factor EB-MuRF-1 axis58but also by modulating SC function.
While an SC abundance issue remains unclear in HF,impairment of SC function and myogenic progression could play an important role in driving muscle wasting,and its key effect on proliferation has been identified.In support of this understanding,myoblast determination protein 1 and myogenin mRNA expression were attenuated 3 days post injury in MI-induced HFvs.control mice.57Furthermore,isolated and cultured SCs treated with angiotensin II showed impairments in proliferation as confirmed via bromodeoxyuridine incorporation.57Similarly,in humans,primary cultures of skeletal muscle myoblasts isolated from the VL of 8 HF patients(with reduced ejection fraction)and 8 healthy matched controls showed proliferation kinetics were delayed at 90 h into the growth phase.57,59In addition,mRNA expression of proliferation factors interleukin-6 and tumor necrosis factor receptor 2 were attenuated in myoblasts derived from muscle samples from HF patients with reduced ejection fraction.Therefore,these findings suggest that SC proliferation is impaired in HF,which may attenuate muscle repair and contribute to atrophy.However,because it remains poorly understood whether HF substantially impacts muscle regeneration,further study in this area is warranted.
4.Effects of HF on contractile function
As presented above,muscle fatigue and weakness are key features of HF.These are determined not only by muscle wasting but also by intrinsic fiber dysfunction evident via a reduction in specific force,which is often termed contractile dysfunction and is consequent to impaired excitation-contraction coupling (ECC).60Impaired ECC increases motor unit firing frequency to meet muscular demands,thereby accelerating muscle fatigue and heightening symptoms of ventilation/breathlessness.61It is often underappreciated how the loss of contractile function,which is a major clinical problem in HF patients because it limits their daily activities and quality of life,may also be caused by sarcopenia.62Sarcopenia is regularly used to define the loss of muscle mass and strength associated with aging,63with studies demonstrating that low muscle force production is more predictive of falls than is low lean mass.64,65
One contributing factor to impaired ECC in skeletal muscle is decreased Ca2+homeostasis (Fig.1).In HF,cytosolic Ca2+fluxes during muscle contraction are reduced in both limb muscles66,67and the diaphragm.68Alterations in Ca2+homeostasis have profound effects on muscle performance,and it seems that reduced sarcoplasmic reticulum (SR) release and reuptake both contribute to muscle weakness in HF.Post-MI,HF rats showed prolonged Ca2+transients and reduced SR release in extensor digitorum longus,accompanied by lower twitch and tetanic tension as well as fatigue resistance;69a similar pattern was found in diaphragm fibers.68Sarcoplasmic or endoplasmic reticulum Ca2+ATPase (SERCA) is largely responsible for Ca2+uptake in cardiac and skeletal muscle(predominantly in the adult SERCA1a and neonatal SERCA1b isoforms in skeletal muscle,with SERCA2a also present in slow skeletal muscle70).HF rats have decreased SERCA1a protein expression in limb and respiratory muscle,which likely impairs Ca2+reuptake to blunt ECC.70A similar trend was also observed in the VL of HF patients,whose biopsies presented lower SERCA2a protein content and diminished levels of phosphorylated phospholamban,both of which reduce Ca2+sequestration into the SR.71Interestingly,expression of SERCA1 and SERCA2a were higher in diaphragm biopsies from HF patients when compared to controls with coronary heart disease,indicating a divergence between limb and respiratory muscle.72Strong evidence supports the idea that the impairment of SR Ca2+release dynamics in HF is caused by dysfunction of the ryanodine receptor 1 (RYR1)complex (i.e.,the main channel responsible for SR Ca2+release in skeletal muscle),which also disrupts basal fiber homoestasis.67,73-75For example,binding of FK506 binding protein 12 (FKBP12,also termed calstabin) to RYR1 in order to stabilize the closed state is diminished in HF.74This is partly due to hyperphosphorylation of RYR1 by protein kinase A due to chronic b-adrenergic signaling,75which promotes Ca2+leakage from the RYR1 into the cytoplasm.VL biopsies from HF patients demonstrate hyper phosphorylation of RYR1 and depleted FKBP12 binding76as well as lower 1,4 dihydopyridine receptor (DHPR) protein content,71while the diaphragm in HF patients also showed lower incidence of FKBP12 binding to the RYR1 complex.72In addition,sensitivity of single diaphragm muscle fibers to cytoplasmic Ca2+concentrations is decreased in experimental HF,while single muscle fibers in HF patients demonstrate reduced actomyosin ATPase activity regardless of fiber type.77These conditions likely combine to further reduce contractile function.78Other factors that play a role include reduced contractile protein content,per se,and the associated post-translational oxidative modifications,with increased nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondrial ROS identified as contributing to diaphragm dysfunction during experimental HF.79,80Studies have further determined that neuromuscular junction (NMJ) fragmentation occurs in HF,81as it does in aging,82but it remains unclear whether this reduces muscle function.83
Disruptions to Ca2+homeostasis in HF may also exacerbate atrophy in both limb and respiratory muscle.In an experimental HF model,greater calpain activity has been found in limb muscle84alongside raised resting Ca2+levels in atrophied diaphragm muscle.85High cytosolic Ca2+concentrations can activate calpains in skeletal muscle,86which may accelerate proteolytic activity via UPS to drive fiber atrophy.60,84Calpains also induce sarcomere disorganization through the degradation of the Z-disk,leading to a reduction in isometric force.Calpain inhibition preserves sarcomere structure,indicating the key role of calpains in contractile dysfunction.87Furthermore,greater cytosolic Ca2+concentration leads to increased ROS production from the key sources,including mitochondria,NADPH oxidase (Nox),and xanthine oxidase,in both limb and diaphragm muscle in experimental HF.60,72,80,88-90Upregulation of Nox has been found in diaphragm biopsies of HF patients,alongside greater protein oxidation,in spite of increases in antioxidant enzymes.79This increase in ROS can lead to the upregulation of key catabolic factors,such as E3-ligases,resulting in muscle atrophy and post-translational oxidative modifications of sarcomeric proteins,which contribute to impaired function.60Targeting these sources of ROS may prove beneficial in the treatment of exercise intolerance in HF,a notion that is supported by various studies.For example,reduction in mitochondrial ROS through the use of a mitochondrial-targeted antioxidant80and a neutral sphingomyelinase inhibitor91preserved diaphragm dysfunction in HF rats post MI.Additionally,inhibition of xanthine oxidase in mice with HF prevented the atrophy of type I and type II fibers92in limb muscle and preserved exercise capacity.Interestingly,only certain isoforms of Nox seem to play a role in diaphragm abnormalities in HF;knockout of a subunit necessary for Nox2 activity restored diaphragm function,88whereas Nox4 knockout had no impact on acute MI.93While complex,ROS may also facilitate the dissociation of FKBP12 from the RYR1,destabilizing the closed state and perpetuating further Ca2+leaks.94,95
Muscle force production is affected by mitochondrial dysfunction and oxidative stress.However,the underlying mechanisms by which oxidative stress contributes to HFrelated muscle wasting remain poorly understood.The role of chronic oxidative stress in a mouse model lacking the antioxidant enzyme copper-zinc-superoxide dismutase shows a progressive decline in mitochondrial function and an increase in ROS production caused muscle atrophy.96When aged mice were evaluated,a striking increase in muscle mitochondrial content near the NMJs was found.However,the function of mitochondria was impaired and an increase in denervated NMJs leading to a reduction in force production was observed.This study suggested that NMJ degeneration and mitochondrial dysfunction are potential mechanisms of sarcopenia.96
Given the greater prevalence of HF in older people97and the negative effects of aging on skeletal muscle,quantifying the independent contribution of agingvs.HF to skeletal muscle dysfunction is a complex task.In aging,uncoupling of DHPR and RYR1 occurs,and Ca2+spark duration is reduced,both of which likely contribute to a reduction in specific force generation.98,99Similarly,HF risk is increased with sedentary behavior,and exercise intolerance may limit physical activity in HF patients,100again complicating the roles of HF and inactivity in muscle dysfunction.Indeed,inactivity has been found to decrease specific force in young humans and old rats.101,102Therefore,it is likely that abnormal Ca2+homeostasis and mitochondrial dysfunction in HF collectively contribute to weakness not only via intrinsic fiber dysfunction but also by promoting fiber atrophy.
5.The effects of exercise training on HF-induced muscle wasting and dysfunction
In the past few years,research groups worldwide have tried to uncover ways to prevent chronic disease-related muscle wasting and dysfunction.Numerous pharmacological and nonpharmacological interventions have been tested,but they have shown limited efficacy.103Therefore,a combination of interventions emphasizing the importance of a healthy lifestyle,diet,and physical activity have been proposed.Exercise training,specifically aerobic exercise training (AET),is associated with improved quality of life,reduced hospitalizations,and prolonged survival104and should be considered an adjuvant therapy to counteract muscle defects in HF.
AET can act in a preventive and/or therapeutic way for a number of non-communicable chronic diseases.105,106Among several abnormalities observed in HF,one of the main features is early muscle fatigue leading to exercise intolerance,and this is related to reduced peak oxygen consumption.107,108In fact,lower aerobic capacity is strongly related with precocious death in healthy subjects and those with cardiovascular disease.109More than a decade ago,high-intensity AET was proposed as an alternative to moderate-intensity AET for stimulating higher levels of peak oxygen consumption in HF patients,110but the effects of both AET protocols on muscle indices are similar.111Therefore,while AET plays an important role as an adjuvant therapy for counteracting skeletal muscle defects,an intriguing question remains whether RET might be a more effective strategy.Therefore,we will briefly review how AET and RET could benefit HF patients by impacting muscle mass,regeneration,and function,as summarized in Fig.1.
5.1.Muscle mass
Protein synthesis is essential for maintaining muscle mass,and this seems to be modulated by exercise training in HF.Previous studies showed that 8 weeks of moderate AET(treadmill) activated the Akt/mTORC1 signaling pathway to counteract muscle wasting in an experimental model of HF.12The same type of exercise modulated that pathway in VL muscle samples from patients (e.g.,IGF-1 expression was higher 6 months after training).112
It has been widely reported that the HF-related overactivation of UPS in skeletal muscle is due to increased oxidative stress levels.113-115In HF patients and animal models,AET has been found to induce anti-inflammatory effects in addition to improving antioxidant defenses,mainly by reducing the pro-inflammatory cytokines of tumor necrosis factor-a and interleukin-6 muscle expression116,117and by increasing glutathione peroxidase 1 and catalase enzyme activities.117It was also shown that MuRF-1 expression decreased after 12 weeks of AET in HF patients,which was strongly correlated with lower proteasome activity and decreased myofiber size compared to non-trained HF patients.22,118In addition,moderate AET has been shown to help re-establish proteasome homeostasis to attenuate muscle wasting in both animal models and patients.119Regarding other key proteolytic systems,such as autophagy,further studies will be necessary to clarify the impact of AET on HF.
Previously,RET was avoided by cardiac patients because it was considered to be a potential cause of adverse ventricular remodeling due to high-pressure loads during weightlifting.120However,evidence from the past 2 decades points to the contrary,and recommends RET (Fig.1) across a range of clinical populations.121,122Indeed,RET provides many beneficial effects not only in terms of muscle strength and function,123but in terms of overall full-body mobility124and mental health125as well.HF patients may also experience skeletal muscle hypertrophy at the whole-muscle level126although a lack of evidence remains available to firmly support this suggestion (especially at the myofiber level) indicative of anabolic resistance.127-129In an MI model,4 weeks of RET was found to restore limb muscle weight (relative to body mass) and muscle fiber area to that of sham operated animals.130This was associated with the reduction of MuRF-1 and muscle atrophy F-box mRNA expression to control levels,decreases in myostatin protein expression,and increases in factors associated with muscle growth.130Interestingly,AET also restored muscle mass and fiber area in the same study,and both RET and AET were able to re-establish antioxidant capacity and then reduce oxidative stress.130Similarly,it was found that high-and moderate-intensity AET restored cross sectional area,mitochondrial function,antioxidant activity,and reversed proteolytic signaling in an MI experimental model.24Likewise,maintaining mitochondrial function through targeted anti-oxidant treatment prevented immobilization-induced limb muscle atrophy.131Collectively,therefore,these studies suggest that both aerobic and resistance exercise may prevent atrophy by reducing oxidative stress,in turn blunting catabolic signaling.132
5.2.Contractile dysfunction
While most studies have focused on AET in HF,a study where HF patients performed 18 weeks of RET showed improvement in muscle strength despite a lack of myofiber hypertrophy.31This could have resulted from improvements in force production for a given level of Ca2+,as is seen with aging.133Thus,it is important to realize that RET or AET may be of benefit to the contractile function of muscle in HF patients independent of muscle mass gains.Alternatively,blood flow restriction exercise in the form of resistance exercise and aerobic exercise134showed benefits (e.g.,in functional capacity,isometric strength,endurance,and quality of life) in HF patients after 6 weeks without concomitant increases in mass.135
The mechanisms by which exercise training benefits contractile function in HF are still being revealed,but one of them appears to be related to HF-induced NMJ fragmentation,which has negative effects on muscle mass.81AET has been shown to reduce the proportion of fragmented NMJs in aged mice,136and as such,this type of exercise may play a role in preserving muscle mass in HF through the same mechanism.
Another probable mechanism is related to the HF-induced Ca2+dysfunction of myofibers.It is known that exercise training increases expression of DHPR,RYR1,and SERCA proteins,137with experimental models suggesting a link between improved exercise tolerance in HF with AET and restored expression of Ca2+-related proteins,in particular DHPR,RYR1,SERCA1,and SERCA2.138Studies have shown that restoring Ca2+homeostasis in skeletal muscle in HF may be achievable via pharmacological treatment that mimics exercise-related benefits.For example,the use of RYR1 stabilizing agent S107 improves exercise tolerance in diaphragm139and limb muscles94by reducing Ca2+leaks through improved FKBP12 binding.A rat model of HF using abdominal aortic coarctation improved limb muscle fatigue resistance and perfusion after 4 weeks of AET(voluntary wheel running);these same markers also improved in rats subject to 2 weeks of overload (a potent angiogenic stimulus akin to RET).140This suggests a close link between peripheral vascular and contractile function in HF.Improved mitochondrial function also occurs after AET (and blood flow restriction exercise in the form of resistance exercise135),likely improving oxidative capacity to enhance fatigue resistance while reducing ROS production to alleviate associated myofilament damage.Reducing ROS production through exercise may result in reductions in mitochondrial ROS through the use of a mitochondrialtargeted antioxidant-maintained maximal specific force in the diaphragm of mice with experimental HF.80
HF is also associated with respiratory muscle weakness,and the effects of both AET and inspiratory muscle training in HF patients were investigated by this.It showed that both protocols are safe and effective in HF for improving quality of life and enhancing muscle mass,leg blood flow,and overall functional capacity.141More direct studies assessing fiber contractile function in the diaphragm have used HF experimental models.For example,9 weeks of AET prevented diaphragm dysfunction in post-MI HF mice,and such effects were associated with attenuated proteolytic pathway expression (UPS and calpain) and oxidative contractile protein modifications (actin and creatine kinase),likely via the upregulation of antioxidant enzyme expression.60Similar findings have been reported in pre-HF animal models of hypertension where 4 weeks of high intensity interval training prevented diaphragm dysfunction.142
5.3.Satellite cells
Limited evidence exists connecting the effects of exercise training and SC dynamics in HF.SCs are activated in response to exercise,which is concomitant with an increase in gene transcription of myogenic regulatory factors,143-146and an increase in SC content is typically observed.147In line with this,endurance exercise training has been shown to alleviate declines in SC content as well as impairments in proliferation and differentiation capacity in aged rodents.148,149Running performance is also positively correlated with SC content in the rats’muscle.150Whether similar exercise interventions can alleviate SC impairments and thus exercise intolerance in HF is unknown.One study proposed the importance of myofiber capillarization for the SC response to RET-induced muscle hypertrophy;thus,healthy young men and women underwent aerobic conditioning for 6 weeks followed by 10 weeks of RET in order to investigate how prior aerobic conditioning alters SC content,activity,and myofiber hypertrophy.151Those with the greatest capillary-to-fiber perimeter exchange index before RET had the greatest change in muscle hypertrophy.Importantly,SC content,activation,and differentiation increased more in the Type I myofiber,which may in part be modulated by enhanced capillarity given the close relationship between the SC and the endothelial niche.151Moreover,baseline capillarization has been found to be predictive of hypertrophic response in older people,152,153who are thought to demonstrate anabolic resistance.154HF patients are well known to have reduced capillarity,and a link between blunted hypertrophy and lower capillarization was shown in experimental HF rat models.140This suggests that aerobic conditioning prior to RET can improve muscle adaption by increasing capillarization,thus reinforcing the idea that engagement in a regular exercise training program involving both aerobic and strength conditioning can be a reliable strategy to counteract HF-induced muscle wasting and dysfunction.
6.Contribution of aging and physical inactivity to the skeletal muscle phenotype in HF
Given the greater prevalence of HF in older people97and the negative effects of aging on skeletal muscle,it is difficult to separate out the contribution of HFper seto skeletal muscle dysfunction.Indeed,in both aging and HF,the diaphragm seems to demonstrate a reduction in fiber cross-sectional area85,155(although this isn’t always true in HF78).Similarly,the isometric force of limb and diaphragm muscle is decreased in both aging(i.e.,sarcopenia)98,99and HF,78but reductions in myofibrillar protein content do not account for all impairments in function.156Interestingly,skeletal muscle fatigue resistance is maintained157or improved with age but impaired in HF,158and unfortunately,HF risk is increased with sedentary behavior even while exercise intolerance limits the physical activity of HF patients.100However,while many of the symptoms of HF may be attributable to inactivity,159a number of studies have confirmed the effects of HF are independent of inactivity160,161and age.For example,most animal studies of HF use young animals,who still develop muscle dysfunction,and data indicate muscle alterations are induced independent of age,with young and old patients responding similarly to exercise training.22Therefore,some but not all muscle alterations in HF can be explained by disuse and aging,which clearly indicates the existence of a muscle pathology.
7.Conclusion and future perspectives
In this review,we have demonstrated promising progress in understanding the basic mechanisms that underpin perturbed skeletal muscle health in HF.This knowledge is pertinent given that HF is one of the most common causes of hospitalization15and that low skeletal muscle mass is an independent risk factor for mortality in HF.162The problem is compounded in societies with aging populations as HF is more prevalent in older people,163of whom 10% are estimated to have sarcopenia.164The mechanisms involved include impairments in SC proliferation,anabolic-catabolic signaling,and myofiber calcium homeostasis.Importantly,we have also shown that exercise,particularly AET,attenuates a number of these impairments in the context of HF(Fig.1).Despite this,future research is required to investigate the specific role played by SCs in skeletal muscle dysfunction.Moreover,while it is well established that exercise can reverse some skeletal muscle deficits in HF,we have a poor understanding of how this is achieved,which limits the potential benefits of exercise prescription.Greater scientific understanding of the mechanisms by which exercise improves skeletal muscle health in HF would provide targets for pharmacological mimetics for bedridden patients unable to perform physical activity,which at present can only provide limited benefit.165
Acknowledgments
This work was supported by Heart Research UK (Grant number 119191) and British Heart Foundation (Grant number 124055).Fig.1 was created using BioRender.com (https://www.biorender.com/).
Authors’contributions
HG,PWH,MGP,and TSB conceived,planned,drafted,edited,and revised the manuscript.All authors have read and approved the final version of the manuscript,and agree with the order of presentation of the authors.
Data access statement
The authors declare that no original data are associated with this article.
Competing interests
The authors declare that they have no competing interests.
 Journal of Sport and Health Science2023年5期
Journal of Sport and Health Science2023年5期
- Journal of Sport and Health Science的其它文章
- How to make the “jump” on understanding the importance of the intrinsic foot muscles for propulsion
- Public health research on physical activity and COVID-19:Progress and updated priorities
- Examining the intrinsic foot muscles’capacity to modulate plantar flexor gearing and ankle joint contributions to propulsion in vertical jumping
- Exploring overweight and obesity beyond body mass index:A body composition analysis in people with and without patellofemoral pain
- Machine-learning-based head impact subtyping based on the spectral densities of the measurable head kinematics
- The management of the long head of the biceps in rotator cuff repair:A comparative study of high vs.subpectoral tenodesis