
碱基编辑技术及其在微生物合成生物学中的应用
2023-09-16 03:06:02王雁南孙宇辉
合成生物学 2023年4期
王雁南,孙宇辉
(武汉大学药学院,组合生物合成与新药发现教育部重点实验室,湖北 武汉 430071)

自DNA 双螺旋结构发现和重组DNA 技术发明以来,人们对于如何高效地实现基因的精准分析和定向改造的探索就不曾停止过。基于CRISPR/Cas 系统开发的一系列基因编辑工具,因其灵活、高效、普适的特点得到了非常广泛的应用,2020年诺贝尔化学奖授予对CRISPR/Cas 系统的发现和发展做出重要贡献的Emmanuelle Charpentier 和Jennifer Doudna,更使CRISPR/Cas 系统及其相关技术受到了人们的瞩目和厚望。
原核生物的CRISPR/Cas 系统是一类可遗传的适应性免疫系统,它可以存储原核生物曾被感染的信息,当再次被感染时,可利用RNA 引导的核酸酶破坏噬菌体遗传物质,从而发挥防御或免疫功能。人们将该系统中成簇规律间隔的短回文重复序列命名为CRISPR(clustered regularly interspaced short palindromic repeats)序列,而将一些邻近CRISPR 序列且具有特征性的相关基因命名为Cas(CRISPR‑associated gene)[1]。CRISPR/Cas 系统被用作基因编辑工具时,通常由单链引导RNA(single guide RNA, sgRNA)引导Cas 蛋白结合在目标位点并产生双链断裂(double strand break,DSB),随后经细胞的同源修复(homology‑directed repair, HDR)介导精准编辑或非同源末端连接修复(nonhomologous end‑joining, NHEJ)引入插入和缺失(insertions and deletions, Indels)等突变,从而实现对目标序列的靶向编辑[2‑3]。尽管CRISPR/Cas 系统相比于以往基因编辑技术,已经展现出巨大优势和成功,但人们发现CRISPR/Cas 系统在使用时仍存在不少问题,例如在对人类遗传疾病中最常见的遗传变异--点突变[4]进行编辑修复或是构建遗传疾病模型时,依靠HDR 进行修复的策略编辑效率较低,并且在应用时还存在一系列的限制条件。此外,Cas 蛋白可能会在脱靶位点产生双链断裂,甚至目标位点DNA 修复途径的活化都可能对细胞产生不利影响[5‑6]。因此,一种无需引入双链断裂即可实现碱基转换的基因编辑工具--碱基编辑器(base editor, BE)应运而生[7]。该工具通过将胞苷或腺苷脱氨酶以及其他功能元件与失去双链切割活性的Cas 蛋白相融合,由sgRNA引导,实现对基因组上目标位置的胞嘧啶或腺嘌呤的碱基转换。碱基编辑技术一经问世,便在生物学、医学等领域展现出巨大的应用潜力。
合成生物学是近些年发展日益蓬勃的交叉学科,它将工程原理应用于对生命系统的研究和改造,以期对细胞行为进行创建、编程和控制。合成生物学在过去数十年中,在化工、能源、材料、农业、环境和医药等领域已展露出巨大的生命力。随着合成生物学的发展,越来越多的化合物可以通过改造培养微生物获得,人们也在不断追求高效精准地改造微生物的能力。而CRISPR/Cas 系统的人工靶向可设计性等优点受到了合成生物学领域研究者的青睐,基于CRISPR/Cas 系统开发的碱基编辑技术在保留上述CRISPR/Cas 系统优点的同时,避免了对目标位点DNA 造成双链断裂,降低了基因编辑产生的细胞毒性。因此,本文对目前已开发的几类主要的DNA 碱基编辑器的发展和工作原理进行了阐述,并重点介绍了碱基编辑器靶向范围受限和拓展措施,以及造成的脱靶编辑的检测和优化两方面内容。同时,本文还介绍了我国部分科研工作者在微生物合成生物学领域运用碱基编辑技术的一些应用,并展望了碱基编辑技术的发展及其在微生物合成生物学领域的应用前景。
1 碱基编辑技术概述
1.1 碱基编辑器的发展
1.1.1 胞嘧啶碱基编辑器和腺嘌呤碱基编辑器的发展
DNA 碱基编辑器最初是由美国哈佛大学David R. Liu 研究团队于2016 年开发的一种基因编辑工具[7],其开发的胞嘧啶碱基编辑器(cytosine base editor, CBE)可以实现碱基对C‑G 到T‑A 的转换。他们将大鼠来源的胞苷脱氨酶APOBEC1 通过一段XTEN linker 连接在dCas9(含D10A 和H840A 突变)的N 端,得到的第一代碱基编辑器BE1通过sgRNA引导结合在目标位点,由dCas9打开双链DNA形成DNA:RNA R‑loop结构。胞苷脱氨酶对暴露出的单链DNA 上局部的胞嘧啶脱氨,形成尿嘧啶。U·G 碱基对随后经过体内DNA 修复或复制依次形成U·A、T‑A 碱基对,即在DNA 非靶标链上4~8 号位的窗口内(将NGG PAM 所在位置定义为21~23,下同)实现了C 到T 的转换。较低的编辑效率可能是由于体内的尿嘧啶DNA 糖基化酶(uracil DNA glycosylase, UDG)切除了脱氨形成的U,使得DNA 被修复回C‑G 碱基对。于是该团队将抑制UDG活性的尿嘧啶DNA糖基化酶抑制剂(uracil DNA glycosylase inhibitor, UGI)连接于BE1 中dCas9 的C 端,得到的第二代碱基编辑器BE2 编辑活性有了明显的提升。而第三代碱基编辑器BE3 则是将BE2 中的dCas9 替换为nCas9(D10A),通过在DNA 靶标链上产生切刻,使DNA 以发生脱氨后的DNA 非靶标链为模板进行修复,进一步提升了碱基编辑的效率,而且仅产生了1.1%的Indels突变(表1)。
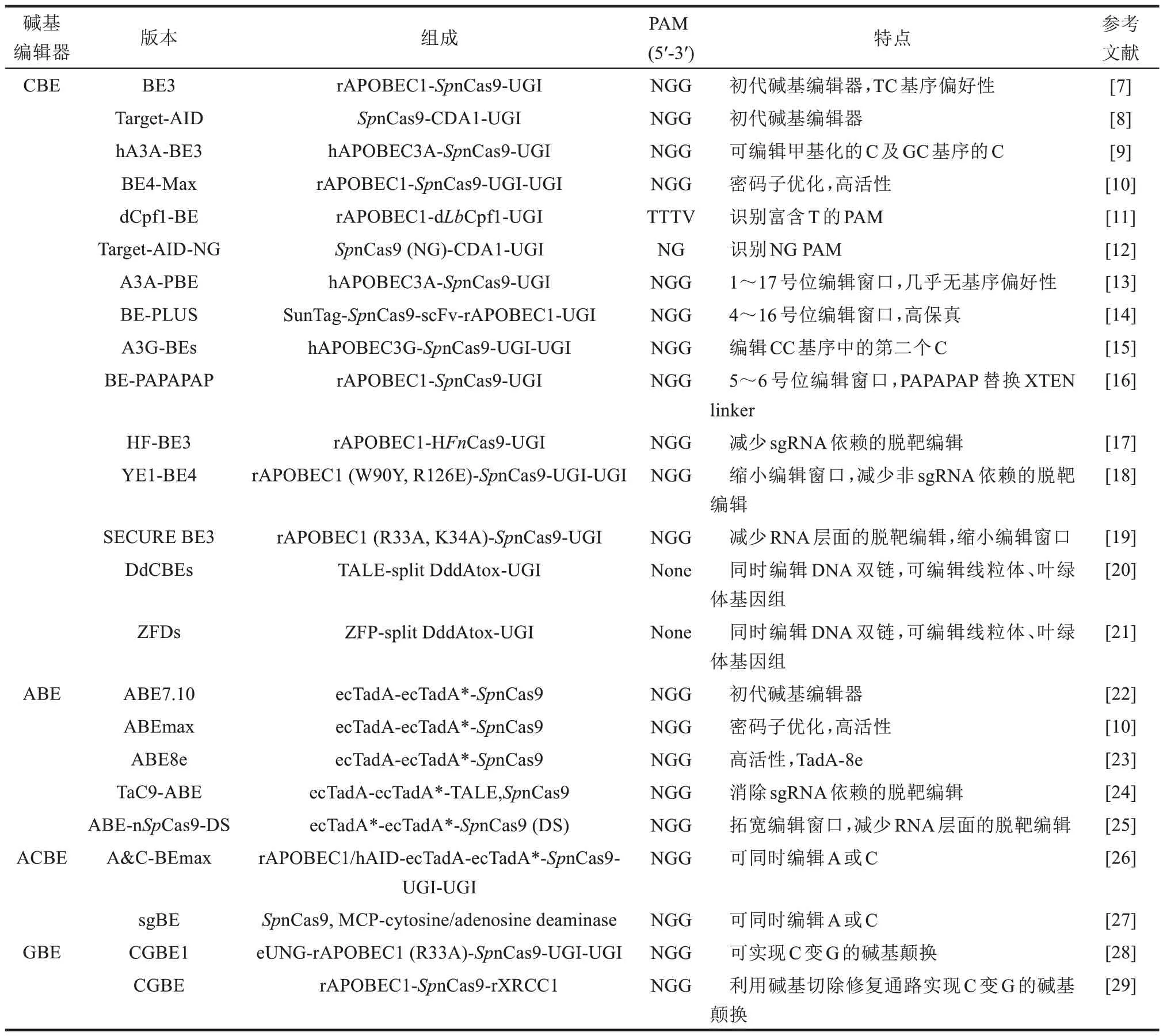
表1 代表性碱基编辑器比较Table 1 Comparison of representative base editors
随后,David R. Liu研究团队[30]通过更换BE3各功能元件之间的linker,并在UGI 的C 端再连接一个UGI,得到的第四代碱基编辑器BE4进一步提高了碱基编辑的效率。在BE4 的N 端连接来自噬菌体Mu 的Gam 蛋白,利用Gam 蛋白结合双链断裂的DNA 末端,使DNA 免于降解,BE4‑Gam 相比BE4降低了Indels突变的产生频率,并且BE4和BE4‑Gam 均提高了C 转换为T 的产物纯度。此外,他们还通过在BE4 两端加上核定位信号,并对BE4 进行密码子优化及祖先序列重构(ancestral reconstruction),由此获得的BE4max 和AncBE4 max 成为目前编辑效率最高的CBE 之一[10]。巧合的是,同在2016 年,日本神户大学Akihiko Kondo研究团队[8]也报道了他们研发的Target‑AID(activation‑induced cytidine deaminase)胞嘧啶碱基编辑器,由nCas9 (D10A)的C 端连接七鳃鳗来源的胞苷脱氨酶(Petromyzon marinuscytidine deaminase 1,PmCDA1)和UGI 组成,其编辑窗口相比于BE3更靠近PAM远端的2~4号位。
2017 年,David R. Liu 研究团队[22]进一步报道了可以实现碱基对A‑T到G‑C转换的腺嘌呤碱基编辑器(adenine base editor, ABE)。与CBE 不同,自然界未发现以DNA 为底物的腺苷脱氨酶,于是该研究团队通过定向进化和蛋白质工程使大肠杆菌tRNA 腺苷脱氨酶(Escherichia colitRNA adenosine deaminase,ecTadA)克服了这一限制,利用二聚的TadA 与nCas9(D10A)相连接,并通过一系列优化,获得了腺嘌呤碱基编辑器ABE7.10(wtTadA‑TadA*‑nCas9,*表示蛋白变体)。ABE7.10在sgRNA 引导下结合在目标位点,由nCas9 打开双链DNA,二聚的TadA 对暴露的单链DNA 上局部的腺嘌呤脱氨,形成次黄嘌呤I,经过体内的错配修复和复制过程,I·T 碱基对依次转换为I·C,G‑C 碱基对,实现在DNA 非靶标链上4~7 号位编辑窗口内A到G的转换,产物纯度极高,且该过程中Indels 突变的发生频率几乎可以忽略不计。之后,David R. Liu 研究团队[10]一直在设法提高ABE 的编辑效率,通过在ABE7.10 两端添加核定位信号和密码子优化,获得了活性更强的ABEmax。进一步通过对TadA‑7.10 使用噬菌体辅助进化方法进行优化,获得的ABE8e 催化速率相比ABE7.10提高了590倍,显示出编辑活性以及与各种Cas蛋白变体和同系物兼容性的大幅提升,由此带来更多的脱靶编辑也可以通过在TadA‑8e中引入V106W突变来加以改善[23]。
1.1.2 其他碱基编辑器的发展
随着CBE 和ABE 相继问世,越来越多的碱基编辑需求寄望于依靠碱基编辑器来实现,但是CBE和ABE仅能实现在目标位点C‑G到T‑A或A‑T到G‑C 的碱基对转换,无法实现其他碱基之间的转变。于是人们自然想到通过将胞苷脱氨酶和腺苷脱氨酶一起融合在nCas9的同侧或两侧,获得的ACBE(fusion adenine and cytocine base editor)可以同时实现目标位点C‑G 到T‑A 和A‑T 到G‑C 碱基对的转换[26,31‑35]。人们将融合了胞苷脱氨酶和腺苷脱氨酶的版本ACBE在哺乳动物细胞和植物细胞中进行了测试,并对系统中的其他因素,例如蛋白之间的linker 及sgRNA 的长度进行了系统的考察,发现这些ACBE都具有较低的脱靶活性。除了将两个不同的脱氨酶与nCas9融合的策略,也有研究团队利用普通版本的CBE 或ABE 并通过在sgRNA 上引入MS2 RNA 适配体招募另一种脱氨酶的策略,也可以实现ACBE 的相似功能[27]。这些ACBE 丰富了目标位点碱基转换的产物种类,在遗传疾病的治疗、细胞谱系追踪以及基因随机突变领域都具有巨大的应用前景[26,31‑35]。
在碱基编辑器的研究和应用过程中,人们发现CBE 和ABE 不仅仅介导C 到T 或A 到G 的碱基转换,还可能会引起C 到G 或C 到A 的碱基颠换[36‑38]。通过分析,发现这一现象可能由于C脱氨后形成的U 被UDG 切除形成AP 位点(apurinic/apyrimidinic site)后,经历细胞内的碱基切除修复过程而导致。于是人们利用UDG 替代CBE 中的UGI,促进细胞切除C 脱氨形成的U,提高了C 碱基颠换的效率。由此开发的GBE(glycosylase base editors)和CGBE(C‑to‑G base editors)可以实现哺乳动物细胞内C 到G[28,39],以及大肠杆菌中C 到A 的碱基颠换[39]。此外,有研究利用参与碱基切除修复途径的rXRCC1蛋白,以及改善影响C 到G 转变过程的其他因素,开发了不同版本的CGBE[29,40‑41]。虽然相继有这一类碱基编辑器的开发被报道,但是细胞内C到G的转变过程并未揭示清晰,这也使得该碱基编辑器在研究和使用过程中遇到的一些问题还难以解答,如GBE 在哺乳动物细胞和大肠杆菌中产生了不同的编辑结果[39]。不过随着该过程的机制被逐渐阐明,更多更高效的CGBE版本有望被开发出来。
此外,人们还开发了可以同时编辑DNA 双链的碱基编辑器:David R. Liu 研究团队通过将能对DNA双链中的胞嘧啶进行脱氨的细菌间毒素DddA蛋白拆分成两个部分,并分别与靶向定位作用的转录激活因子样效应物(transcription activator‑like effector, TALE)或者不同的Cas 蛋白以及UGI 融合,可以实现当两个定位蛋白在DNA 上结合位置相距较近时,聚合的DddA 可以对两者之间的DNA 双链上的C 同时进行脱氨。而其他情况下,两个拆分的DddA 无法在体内聚合,大大降低了该编辑工具的细胞毒性[20]。其中,将拆分的DddA与两个TALE融合得到的碱基编辑器DdCBEs(RNA‑free DddA‑derived cytosine base editors),因无需RNA 引导便可发挥作用,使其可以对无法通过CRISPR/Cas系统进行编辑的线粒体DNA 和叶绿体DNA实现有效的编辑[42]。但是,人们通过全基因组测序方法检测到DdCBE 会在细胞核内造成大量的DNA脱靶编辑[43]。而将分裂的DddA与两个不同的Cas蛋白[如dSpCas9和SaKKH‑Cas9(D10A)]融合之后形成的编辑器,由于系统过于繁复,反而使得其应用变得有限。韩国首尔国立大学Jin‑Soo Kim 研究团队[21]也发表了类似的工作,他们开发的锌指脱氨酶(zinc finger deaminase, ZFD)将分裂的DddA 与锌指DNA 结合蛋白(zinc‑finger DNA binding protein)相融合,也能在核DNA和线粒体DNA 上实现类似的编辑。之后,Jin‑Soo Kim研究团队[44]在DdCBE 的基础上结合腺苷脱氨酶TadA,经历一系列优化改造,开发出了可以在线粒体基因组实现A 到G 转化的碱基编辑器TALED(TALE‑linked deaminase)。
1.1.3 RNA碱基编辑器的发展
除了DNA 碱基编辑器,人们也开发了一系列RNA 碱基编辑器来避免DNA 编辑所带来的遗传信息不可逆改变以及安全与伦理问题。RNA 碱基编辑使用的脱氨酶大多来自ADAR (adenosine deaminases that act on RNA)家族的腺苷脱氨酶,ADAR 包含独特的RNA 结合基序,识别定位双链RNA 的部分区域,ADAR 脱氨酶域(ADAR deaminase domain, ADARDD)将腺苷脱去氨基,实现A 到I 的转变[45‑46]。人们利用引入发夹结构或者化学修饰的反义RNA 与目标RNA 互补,供融合了其他蛋白结合域或标签的ADARDD结合[47‑53],并将需要编辑的腺苷对侧的反义RNA 设计为非互补的胞苷,提高更倾向在A·C 错配处脱氨的ADAR在目标腺苷处的编辑效率[50],这一策略被应用于大多数RNA 碱基编辑器的设计中。而北京大学魏文胜研究团队[54]通过设计ADAR 招募RNA(ADAR‑recruiting RNA, arRNA)来招募细胞内天然的ADAR1 或ADAR2 蛋白,实现目标RNA 从A 到I 的转换。美国哈佛大学张锋研究团队将ADAR2DD与dPspCas13b 融合,通过RNA 引导至目标位点,开发了新的RNA 碱基编辑工具REPAIR(RNA Editing for Programmable A‑to‑I Replacement)。
REPAIR 对报告基因转录产物的编辑效率高于之前通过反义RNA 实现的RNA 碱基编辑,而在细胞内源全转录本的编辑中,其效率也与之前的技术相比更好或持平。为了提升REPAIR 的编辑特异性,张锋研究团队在ADAR2DD引入了E488Q 和T375G两个点突变,开发出了REPAIRv2,大幅减少了全转录组水平和目标腺苷上下游近端的脱靶编辑,但也一定程度上降低了目标位点的编辑效率[55]。他们之后通过对ADAR2DD进行定向进化,开发出了RESCUE (RNA Editing for Specific C to U Exchange)系统,可以实现目标位点RNA 碱基从C 到U 的转换,同时保留了A 到I 转换的活性,仅需通过在目标核苷酸对侧的gRNA中设计不同的错配核苷酸即可实现差异化的编辑[56]。除了ADAR家族的脱氨酶,上海科技大学池天研究团队和中国科学院植物生理生态研究所李轩研究团队基于APOBEC3A 的RNA 的催化活性,构建出可实现C到U 的RNA 碱基编辑器,其中郝沛研究团队还基于晶体结构开发了只具有RNA 催化功能的APOBEC3A(A3A)变体[57‑58]。
1. 2 碱基编辑器编辑范围的拓展
碱基编辑工具在不引入DNA 双链断裂的情况下,实现了高效率的碱基转换(包括颠换),其优异的性能使人们希望碱基编辑器可以适用于更多位点的编辑。碱基编辑器可编辑的位点受限于Cas蛋白的定位以及Cas 蛋白打开DNA 双链之后形成的R‑loop 与脱氨酶相互作用。人们从这两方面入手,开发了一系列具有不同靶向位点和编辑窗口的碱基编辑器。
1.2.1 碱基编辑器靶向位点的拓展
David R. Liu研究团队[59]通过将rAPOBEC1与Cas9 的工程变体融合,获得了可以靶向非NGG PAM 的碱基编辑器SaBE3、Sa(KKH)‑BE3、VQR‑BE3、VRER‑BE3 和EQR‑BE3。2018 年,该研究团队又开发了可识别NG、GAA 和GAT PAM 的xCas9,并考察了它与胞苷脱氨酶和腺苷脱氨酶融合后的编辑效果,发现xCas9 与BE3、ABE7.10(指除了Cas9 蛋白的其余部分)有很好的兼容性,可以靶向它们之前无法编辑的位点,且xCas9(3.7)‑ABE 的编辑效率甚至高于ABE7.10[60]。之后,他们通过定向进化获得了可以识别NRRH、NRTH和NRCH PAM的Cas9变体,结合脱氨酶后,使碱基编辑器可以对PAM 为NR 的位点进行编辑[61]。SpCas9‑NG 和SpRY 也被用于对非NGG PAM的位点进行碱基编辑,研究发现nSpCas9‑NG‑AID(Target‑AID‑NG)的编辑效率要优于xCas9‑BE4[12];而融合SpRY 的碱基编辑器几乎可以独立于PAM 靶向目标位置[62]。此外,人们还开发了基于ScCas9 和SmacCas9 的碱基编辑器,分别识别NNG 和NAAA 序列的PAM[63‑65]。除了利用Cas9 蛋白变体拓展靶向位点,Cas12a 蛋白也被用于脱氨酶的定位。上海科技大学陈佳研究团队[11]通过将rAPOBEC1 与dLbCpf1(dLbCas12a)融合,获得了可以识别富含胸腺嘧啶核苷酸PAM(TTTV)的dCpf1‑BE。美国麻省总医院J. Keith Joung 研究团队[66]通过结构改造获得了可以识别非TTTV PAM的AsCas12a 蛋白变体enAsCas12a,其可以引导胞苷脱氨酶(enAsBE)在非TTTV PAM 的靶点进行碱基编辑。然而,与这些融合Cas9 变体的CBE 不同,使用Cas9 蛋白变体构建的ABE 与经典的ABE版本(ABE7.10)相比,大部分的碱基编辑效率较低,可能是因为TadA 蛋白最初是基于与SpCas9 融合后共同进化而来。此外,美国Beam Therapeutics公司Giuseppe Ciaramella 研究团队[67]也通过基于ABE7.10 的进化获得了TadA*8,其融合SpCas9‑NG和SaCas9展现了较高的编辑活性。
1.2.2 碱基编辑器编辑窗口的拓展
通过更换一些碱基编辑器的Cas蛋白,也可以拓宽碱基编辑器的编辑窗口。如SaABEmax 和SaKKH‑ABEmax 编辑器,相较ABEmax(SpCas9)将编辑窗口扩大为4~14 号位[68]。David R. Liu 研究团队[69]通过将脱氨酶与不同方式循环排列的SpCas9(Cas9 circular permutants, Cas9‑CPs)相融合,试图使脱氨酶更容易接触到单链DNA底物。融合获得的两个CP‑CBEmax 保持了与CBEmax 一致的编辑活性,而将编辑窗口拓展为4~11 号位,并且同时提高了C到T的产物纯度。这一结果印证了循环排列的SpCas9 的新末端使融合的脱氨酶和UGI 更容易接触到单链DNA 底物,从而扩大编辑窗口并阻止UDG 切除U 的推测。而融合后的CP‑ABEmax 将编辑窗口扩大到了4~12 号位,还有一例虽未扩大编辑窗口,但是显示出编辑窗口的平移,并且这些CP‑CBEmax 和CP‑ABEmax 造成的Indels突变比例均有所下降[68]。
除了可以通过更换Cas 蛋白靶向更多的位点,也可以通过更换脱氨酶拓展碱基编辑器的编辑窗口,并改变碱基编辑的序列偏好性。中国科学院遗传与发育生物学研究所高彩霞研究团队[13]通过用APOBEC3A 替换APOBEC1,将碱基编辑器的编辑窗口扩大到1~17 号位,并且有效地解决了APOBEC1 偏好编辑T 下游的C(5'‑TC‑3'基序偏好性),而对G 下游的C(GC 基序)编辑效率低的问题,A3A‑PBE 碱基编辑器对C 上下游序列几乎没有选择性。融合APOBEC3A 的碱基编辑器也弥补了APOBEC1 脱氨酶无法对甲基化的C 进行编辑的缺陷[9]。David R. Liu 研究团队[70]通过用CDA1 替换APOBEC1,并经过密码子优化构建了CDA1‑BE4max,将编辑窗口扩大到1~9 号位。将其进一步定向进化获得的evoCDA1‑BE4max 将编辑窗口扩大到1~13 号位,其与用类似方式优化获得的另外两个碱基编辑器evoAPOBEC1‑BE4max 和evoFERNY‑BE4max 均对GC 基序中的C 展现良好的编辑活性。美国莱斯大学高雪研究团队[15]通过利用仅保留C 端结构域的APOBEC3G(A3G)替换APOBEC1,并通过蛋白质工程改造,获得了A3G‑BE4.4、A3G‑BE5.13 和A3G‑BE5.14 三个版本的碱基编辑器,将编辑窗口扩大到约4~15 号位,并且在编辑底物为CC 基序的情况下只编辑第二个C,大幅减少了对第一个C 的编辑。上海科技大学黄行许研究团队[71]通过优化人A3G 蛋白,开发出的oA3G‑BE3 对CC 和CCC 基序中下划线所在位置的C的编辑相比BE3更具选择性。吉林大学李占军研究团队[72]通过在AID 脱氨酶中引入突变,融合在nCas9(D10A)的N 端,获得的碱基编辑器eAID‑BE4max将编辑窗口拓展为1~11号位。中国科学院脑科学与智能技术卓越创新中心仇子龙研究团队[73]通过融合nCas9(D10A)及一系列七鳃鳗胞苷脱氨酶开发了一系列编辑窗口位于PAM 远端、近端以及覆盖整个Protospacer 区域的碱基编辑器。
也有人通过改变脱氨酶与DNA 的相互作用来拓展编辑的范围。黄行许研究团队开发了BE‑PLUS 编辑器,利用了包含多个GCN4 多肽的SunTaq 信号放大系统[74],通过将SunTaq 标签融合在nCas9(D10A)的N 端,招募单独表达的scFV‑APOBEC‑UGI‑GB1(scFV 抗体特异性识别GCN4多肽,GB1 为促溶标签,消除蛋白质聚集),可以将编辑窗口拓宽到4~16 号位[14]。华东师范大学李大力研究团队通过在BE4max、A3A‑BE4max 及eA3A‑BE4max[75]三个碱基编辑器的脱氨酶与Cas蛋白之间融合Rad51 蛋白的单链DNA 结合域(single‑stranded DNA‑binding domain, ssDBD),获得的hyBE4max、hyA3A‑BE4max 和heyA3A‑BE4max 三个碱基编辑器相比之前的版本大幅拓宽了编辑窗口[76]。四川大学姚少华研究团队[77]将胞苷脱氨酶嵌合在Cas9 蛋白的PAM 相互作用域(PAM‑interacting domain, PI domain)内,获得了编辑窗口拓展为4~14 号位的碱基编辑器BE‑PIGS。复旦大学程天林研究团队[25]通过将腺苷脱氨酶嵌合于Cas9 内部的特定位点,获得了多个版本的ABE‑nSpCas9‑DS 碱基编辑器,这一系列的碱基编辑器将ABE 编辑窗口拓宽为2~16 号位,并且降低了RNA 脱靶编辑的风险。瑞士苏黎世联邦理工学院Gerald Schwank 研究团队[78]通过用TadA替换Cas9 的HNH 结构域,缩小了整个编辑器的大小,并获得了编辑窗口靠近PAM的碱基编辑器。
此外, Jin‑Soo Kim 研究团队[79]通过在sgRNA 5'端延长2~3 nt,将ABE7.10 的编辑窗口向PAM 远端延伸2~3 nt。类似编辑窗口延伸的现象也在dCas‑CDA‑UL、nCas9(D10A)‑AID 和CRISPR‑CDA‑nCas9‑UGI等碱基编辑器结合5'端延长的sgRNA时被发现[80‑82]。
除了拓宽编辑窗口,许多的碱基编辑场景需要碱基编辑器拥有更小的编辑窗口从而产生更精准的编辑。David R. Liu 研究团队通过引入突变降低rAPOBEC1 的编辑活性及底物结合能力,获得了四种(YE1‑BE3、EE‑BE3、YE2‑BE3 和YEE‑BE3)相比于BE3略微降低了编辑活性但是将编辑窗口范围缩小到1~2 nt 的碱基编辑器,而通过使用截短的sgRNA 未能很好地实现这一点[59]。之后李占军研究团队[83]在YE1 的基础上构建了YFE‑BE4max,同样将编辑窗口缩小到3 nt 并在兔子中实现高效率、低脱靶的编辑。德国马普分子植物生理所Ralph Bock 研究团队[16,84]通过用5~7 个氨基酸的刚性linker(PAPAP/PAPAPAP)替换BE3 中原有的XTEN linker,将编辑窗口缩小到了5~7 号位,而在nSpCas9 的N 端融合截短C 端的CDA1 或A3A,分别使得碱基编辑几乎只发生在3 号位或5~6 号位。J. Keith Joung 研究团队[19]为了减少碱基编辑器引发的RNA 随机突变,开发了两个在脱氨酶中引入突变的碱基编辑器变体--BE3‑R33A和BE3‑R33A/K34A,其在大幅降低RNA 随机突变频率的同时,也将编辑窗口缩小到了5~7 号位。中国科学院天津工业生物技术研究所张学礼研究团队[85]通过设计非完全匹配的sgRNA 增加了碱基编辑器对目标序列单个碱基的编辑比例,并且提高了碱基编辑的效率。
1.3 碱基编辑器编辑特异性的优化
1.3.1 碱基编辑器脱靶编辑的检测
在开发和应用单碱基编辑器的过程中,人们发现单碱基编辑器可以在目标位点及脱靶位点产生多种不需要的编辑副产物。因此,研究者针对性地开发出了一系列具有更高特异性的单碱基编辑器,以改善这些问题。
通用的编辑产物检测方法包括对目标位点,以及对通过软件预测或文献已发表的脱靶位点进行桑格测序(Sanger sequencing)或深度测序(deep sequencing),从而进行定点考察[7],或是通过全基因组测序(whole‑genome sequencing, WGS)了解在全基因组的脱靶情况[80],这些方法都具有各自的局限性。
Jin‑Soo Kim研究团队[86]最早在CBE脱靶编辑的全基因组检测方面取得突破,他们使用BE3ΔUGI在体外对编辑位点脱氨并在DNA 靶标链上产生切刻,之后利用尿嘧啶特异性切除试剂,即一种尿嘧啶DNA 糖基化酶及DNA 糖基化酶‑裂解酶内切酶Ⅷ的混合物,将C脱氨形成的U所在位置的DNA 链切断,形成双链断裂。之后,DNA 样本在末端修复和连接后通过全基因组测序,与参考基因组序列或未经编辑处理的对照基因组测序序列比对,寻找基因组上BE3ΔUGI 的编辑位点,该方法被称为Digenome‑seq(digested‑genome sequencing)。随后,中山大学松阳洲研究团队和Jin‑Soo Kim 研究团队[87‑88]分别开发了针对ABE 脱靶编辑全基因组检测的方法,通过类似的策略,先用ABE7.10 在体外对基因组DNA 编辑并产生切刻,之后用内切酶Ⅴ对DNA 再次进行处理,切断脱氨形成的肌苷3'下游的第二个磷酸二酯键,从而产生双链断裂,经过DNA 末端修复后通过全基因组测序寻找编辑位点。但是这种检测ABE 脱靶位点的方法会遗漏ABE 导致的C 到其他碱基的转变,并且以上几种方法都是检测体外编辑的产物,并不能客观地展示体内真实发生的编辑,于是高彩霞研究团队和中国科学院神经科学研究所杨辉研究团队分别对水稻和小鼠体内的碱基编辑进行了全基因组分析:高彩霞研究团队[89]分别用三种不同的碱基编辑器编辑水稻,并设置只转化碱基编辑器而不转化sgRNA 的对照,之后对编辑植株及野生型植株基因组DNA 进行测序,用野生型植株的测序结果过滤背景突变,便可观测到碱基编辑器所引发的突变;杨辉研究团队[90]在小鼠受精卵分裂为两个细胞的时候,对其中一个细胞注射碱基编辑的相关元件以及荧光蛋白基因,在胚胎成长后的第14.5 天,对胚胎进行消化,通过流式细胞仪对基于荧光蛋白表达分选出发生编辑和未发生编辑的细胞,并对二者进行全基因组测序。对比测序结果,过滤掉未编辑细胞中的背景突变,便可得到发生编辑细胞的突变情况,这种方法被命名为GOTI(genome‑wide off‑target analysis by two‑cell embryo injection)。David R. Liu 研究团队和美国Beam Therapeutics 公司Nicole M. Gaudelli研究团队[18,91]还利用报告系统或者通过其他Cas9蛋白结合DNA 形成R‑loop 结构暴露出单链DNA,分别设计了在细菌和哺乳动物细胞中不需要通过全基因组测序即可检测碱基编辑器脱靶编辑的方法。之后北京大学伊成器研究团队[92]开发了针对胞嘧啶碱基编辑敏感度更高的检测技术Detect‑seq(dU‑detection enabled by C‑to‑T transition during sequencing), 通过利用化学标记和生物素Pulldown 技术,追踪CBE 体内编辑后的基因组产物中的中间体脱氧尿苷来揭示CBE 的编辑组(editome)。
1.3.2 碱基编辑器在目标位点产生的编辑副产物及优化策略
通过上述检测手段,人们发现碱基编辑器造成的非预期编辑包括碱基颠换、旁观者编辑(bystander editing)以及Indels 突变。产生碱基颠换的原因在前文已有介绍,即C脱氨后形成的U被UDG切除,形成AP位点后经历细胞内的碱基切除修复而产生C 到A 或G 的颠换;而ABE 的编辑A的过程中发生碱基颠换则未见报道,可能由于体内从DNA 中切除肌苷的酶的活性或丰度较低,导致A 被编辑后经历碱基切除修复的可能性极小[22]。当编辑窗口内存在多个C 或者A 时,除了目标碱基,周围的碱基也被编辑,这种情况被称为旁观者编辑。虽然在某些情况下旁观者编辑的影响不大,例如由于密码子简并性,旁观者编辑可能只会导致沉默突变,或者导致同类型氨基酸的转变,但是其他情况下,旁观者编辑可能会产生非预期的严重后果。Indels 突变的产生则主要由碱基编辑器中nCas9产生的切刻或是碱基脱氨之后经历碱基切除修复所导致。检测结果显示,碱基编辑后确实有少量的Indels突变发生,并且CBE产生的频率一般要高于ABE[22],但碱基编辑导致Indels 突变产生的机理还未被完全揭示[93]。
针对上述情况,人们采取了一系列的改进措施。为了避免产生碱基颠换,人们在碱基编辑器中引入更多的UGI 来降低碱基颠换的概率[14,30]。为了减少旁观者编辑,人们一方面通过上文介绍的缩小编辑窗口的方法,减少脱氨酶可接触的底物数量。另一方面利用脱氨酶编辑序列的偏好性,使得脱氨酶只编辑某些位置的C,例如改造的A3G几乎只编辑CC 基序中的第二个C[15]。此外,韩国汉阳大学Sangsu Bae 研究团队[94]通过在TadA7.10中引入突变,降低了其对旁观者胞嘧啶脱氨的活性及RNA 编辑的活性。美国莱斯大学Anatoly B.Kolomeisky 等研究团队[95]通过数学建模分析预测目标碱基和旁观者碱基的编辑效率,并据此开发了提高目标碱基与旁观者碱基编辑比的A3A 版本。针对Indels突变,通过使用dCas9[7]或AsCas12a[66]引导脱氨酶,或者利用噬菌体Mu 的Gam 蛋白[30]均可以降低Indels突变的发生频率。
1.3.3 碱基编辑器造成的sgRNA 依赖和非sgRNA依赖的DNA脱靶编辑及优化策略
除了在目标位点产生非预期的编辑外,碱基编辑器脱靶到其他DNA 位点也会产生非预期的编辑产物,包括sgRNA 依赖的DNA 脱靶编辑以及非sgRNA依赖的DNA脱靶编辑。
碱基编辑器所造成的sgRNA 依赖的DNA 脱靶编辑的发生位点并不完全与sgRNA 引导Cas 蛋白所造成的脱靶编辑的位点相同,其中还受到编辑窗口内是否存在脱氨底物、底物上下游的核苷酸种类以及脱氨酶能否接触到R‑loop 结构中DNA 链等因素影响。人们发现这一类脱靶编辑的发生相比非sgRNA 依赖的脱靶编辑概率较小[89],但仍给碱基编辑器的应用带来了潜在的风险,因此人们对该问题进行了一系列的优化。通过使用高保真Cas 蛋白替换野生型Cas 蛋白可以有效降低CBE 和ABE 这一类脱靶编辑发生频率,例如HF‑Cas9,xCas9(3.7)、Sniper‑Cas9、HypaCas9 等[17,60,75,87,96]。中国科学院广州生物医药与健康研究院赖良学与五邑大学邹庆剑研究团队合作将腺苷脱氨酶或胞苷脱氨酶与转录激活因子样效应子融合,并依靠靶向附近的dCas9 打开DNA 双链,开发了TaC9‑ABE 和TaC9‑CBE 碱基编辑器。该系统可以消除sgRNA 依赖的脱靶编辑,而不影响靶向编辑效率[24,97]。有研究显示使用截短的sgRNA 可以在不大幅降低目标位点编辑活性的情况下,减少碱基编辑器sgRNA依赖的DNA脱靶编辑[96],但是也有研究发现使用截短的sgRNA 会导致碱基编辑器在目标位点和脱靶位点的编辑活性均有所降低,并且当sgRNA 与DNA 的错配出现在PAM 远端时,使用截短的sgRNA 反而会展现出更高的脱靶活性[86],表明在碱基编辑器中使用截短sgRNA 来提升特异性的方法并不如在Cas9 中适用[98]。而使用5'端添加1~2 个G 的sgRNA 则可以在不降低目标位点编辑活性的情况下减少sgRNA 依赖的DNA 脱靶编辑[86],但也有研究显示脱靶的减少只在sgRNA 5'端添加2个G的情况下出现[88]。武汉大学孙宇辉研究团队[99]通过在sgRNA 的5'端引入气泡发夹结构,也可以有效地提升碱基编辑器的编辑特异性。
碱基编辑器所造成的非sgRNA 依赖的DNA 脱靶编辑通常由CBE 引发,即使在没有sgRNA 的情况下,CBE 也能引发一定量的非sgRNA 依赖的DNA 脱靶编辑。而在ABE 的编辑产物中几乎没有检测到此类脱靶编辑,发生频率仅接近自发突变。这些突变主要是C到T的单核苷酸变异,被认为是由胞苷脱氨酶和UGI 在整个基因组中长期随机的反应而产生。而DNA 腺苷脱氨酶本身由RNA 腺苷脱氨酶进化而来,与DNA 结合能力较弱,所以ABE 几乎不引发非sgRNA 依赖的DNA 脱靶编辑[89‑90,100]。针对这一问题,David R. Liu 研究团队[18]通过测试, 发现YE1‑BE4、 YE2‑BE4、R33A+K34A‑BE4 等7 个碱基编辑器均减少了DNA层面的各种脱靶问题,其中YE1与其他Cas变体展现了更好的兼容性,可以高效地编辑更多的位点。Nicole M. Gaudelli 研究团队[91]从153 个脱氨酶中筛选出4 个脱氨酶变体--PpABOBEC1、RrA3F、AmAPOBEC1和SsAPOBEC3b,由它们所构建的碱基编辑器降低了非sgRNA 依赖的脱靶编辑活性,并通过对这些脱氨酶进行工程改造,进一步降低了融合这些脱氨酶的碱基编辑器DNA 和RNA 的脱靶编辑频率。杨辉研究团队[101]从23 个理性设计的CBE 变体中筛选出在目标位点保留了高编辑效率,同时引发极低DNA 和RNA 脱靶编辑和旁观者编辑的YE1‑BE3‑FNLS变体。
1.3.4 碱基编辑器造成的RNA 脱靶编辑及优化策略
此外,人们还通过RNA 测序,发现碱基编辑器除了造成DNA 层面的脱靶编辑,还会引起大量的非sgRNA 依赖的RNA 脱靶编辑[19,102‑104],脱靶的原因被认为是CBE 和ABE 脱氨酶均具有对RNA脱氨的活性。而RNA 脱靶编辑除了低水平、广泛而随机地发生在内源RNA 上,也可能造成对sgRNA 和碱基编辑器mRNA 的编辑,进而可能诱发DNA 层面脱靶编辑的发生[19]。针对这些问题,人们通过在脱氨酶中引入突变,构建了多种碱基编辑器变体,减少了RNA脱靶编辑的发生,其中一些突变也减少了DNA脱靶编辑的发生[19,23,67,91,101‑104]。另外,也有研究通过将腺苷脱氨酶嵌合于Cas9 内部的特定位点,限制了腺苷脱氨酶的活性,减少了RNA脱靶编辑的发生[25]。
1.3.5 碱基编辑器特异性优化的其他策略
除了蛋白层面的工程改造,也可以通过时空控制减少碱基编辑器的剂量或作用时间来提升碱基编辑器的特异性。碱基编辑器可以通过核糖核蛋白[17‑18,23,105‑106]、mRNA[61,67,79,107]或逆转录病毒颗粒[108]的方式递送到细胞内。通过这几种方式递送的碱基编辑器在体内都更容易被降解,减少了碱基编辑器在体内的存在时间,从而减少脱靶编辑发生的机会。利用anti‑CRISPR 蛋白AcrⅡA5 可以有效地抑制Cas9的活性[109],利用anti‑deaminase(Ade)蛋白可以有效地抑制脱氨酶APOBEC3A 的活性[110],从而减少碱基编辑器的脱靶。通过在sgRNA 中设计RNA 适体酶(aptamer)来招募脱氨酶编辑可以提升编辑的特异性[111]。此外通过类似于拆分Cas蛋白的方式[112‑113]拆分脱氨酶[114],可以精准地控制编辑的时间,减少额外编辑的发生。陈佳研究团队[115]利用其首次发现的、具有胞苷脱氨酶抑制作用的胞苷脱氨酶的调节结构域调控胞苷脱氨酶,开发了高特异性的碱基编辑器tBEs(transformer base editors)。
2 碱基编辑技术在微生物合成生物学领域的应用
由于碱基编辑器可以在不引入双链DNA 断裂的情况下实现碱基的转换,于是人们可以方便地将碱基编辑器应用于提前终止密码子的引入和修复。如CBE 可以将CAA、CAG、CGA 或TGG 密码子转换为终止密码子TAA、TAG 和TGA 来提前终止基因的表达[116‑117],以及通过ABE 将起始密码子ATG 转变成ACG 扰乱基因翻译的起始,都可以起到类似基因失活的作用[118],用于考察基因的功能或定向改造生物代谢途径。相反,ABE 也可以通过将提前的终止密码子变成谷氨酰胺(CAA、CAG)或精氨酸(CGA)的密码子,使提前终止的基因翻译恢复正常[119]。在以代谢产物为主要研究目标的微生物中,人们对于脱靶所带来的非预期后果的容忍度远高于以人类疾病治疗为目标的研究。因此,碱基编辑技术和策略迅速被运用于微生物合成生物学研究领域,我国科研工作者在许多重要微生物天然产物的生物合成途径研究及改造中一展身手,也为基因编辑技术的发展提供了持续不断的驱动力。例如:
中国科学院分子植物卓越中心杨晟研究团队[120]利用其构建的适用于梭菌Clostridium beijerinckii的胞嘧啶碱基编辑器pCBEclos‑opt,在C. beijerinckiiNCIMB 8052 中使得编码木糖代谢的转录调节因子xylR产生C‑G 至T‑A 的突变,所获得的突变株木糖消耗量比野生型菌株高10%。
北京大学席建中研究团队通过CBE(dCas9‑CDA1‑UL)在基因中引入提前的终止密码子,逐个敲除了红细菌Rhodobacter sphaeroides2.4.1 中参与抗氧化剂辅酶Q10生物合成的系列基因,揭示了这些基因在合成辅酶Q10过程中的重要性。还通过敲除八氢番茄红素合酶基因crtB,以及同时敲除crtB和参与光合氧化还原信号调节的基因ppsR,将野生型中辅酶Q10的产量分别提高了6% 和39%[121],向人们展现出了该技术和策略的实用性。
孙宇辉研究团队[122]构建了适用于链霉菌属的CBE 和ABE,并通过CBE 在Streptomyces avermitilisMA‑4680中olm、rpp、pks1、pks4和pks8五个甚至多达九个PKS 基因簇内同时引入提前终止密码子,提高S. avermitilisMA‑4680 中阿维菌素的产量。之后,该研究团队将其开发的适用于链霉菌的碱基编辑器成功应用于揭示潮霉素B 的生物合成途径。他们利用CBE 在Streptomyces hygroscopicussubsp.hygroscopicusDSM 40578 参与潮霉素B 生物合成的五个候选基因内引入提前的终止密码子或突变编码区内的保守残基序列使基因失活。结合体内外的实验证据揭示了蛋白HygJ、HygL 和HygD 负责核苷二磷酸庚糖的连续脱氢、转氨基和转糖基化,HygY 是一种不同寻常、作用于羟基碳的自由基S‑腺苷甲硫氨酸依赖的差向异构酶,而HygM 是一个在多平行代谢网络中发挥作用的多功能甲基转移酶。这些工作,特别在多基因同步考察上都展现出了碱基编辑器有别于传统方法和策略在天然产物生物合成机制研究领域的独特优越性[123]。
中国科技大学俞汉青研究团队[124]也在希瓦氏菌Shewanella oneidensisMR‑1 中开发了快速且强大的碱基编辑器pCBEso,并应用该工具快速鉴定出S. oneidensisMR‑1 内参与N‑乙酰氨基葡萄糖(GlcNAc)或葡萄糖代谢的关键基因。此外,他们还利用pCBEso 构建了具有扩展碳源利用谱的S.oneidensisMR‑1 工程菌株,当葡萄糖或GlcNAc 成为唯一碳源时,其对多种有机污染物(偶氮染料和有机砷化合物)的降解率高于野生型菌株。
上海交通大学肖毅研究团队[125]针对假单胞菌属的腺嘌呤碱基编辑器开发了一种基于ABE 的蛋白质功能障碍预测方法DABE‑CSP(dysfunctionviaABE through CRISPOR‑SIFT prediction),利用该方法,他们发现了黏康酸(muconic acid)代谢途径中的基因catB失活或catB和catC同时失活会导致黏康酸的积累,并且后者以100%的产率积累黏康酸。DABE‑CSP 方法的建立可以加快对假单胞菌属物种的研究。
上海师范大学芦银华研究团队[126]利用其构建的胞嘧啶碱基编辑器dCas9‑CDA‑ULstr,在链霉菌Streptomyces rapamycinicus2001 内抑制雷帕霉素合成的基因rapS中引入提前终止密码子,使雷帕霉素产量提高了63.3%。
浙江大学吴坚平研究团队[127]利用其构建的适用于假单胞菌属的多重胞嘧啶碱基编辑系统提高了Pseudomonas putidaKT2440 中儿茶酸的产量。他们利用该系统在原儿茶酸3,4‑双加氧酶的β亚基(PcaH)和丙酮酸激酶(PykA)编码基因中引入提前终止密码子,并通过该系统在3‑脱氧‑2‑阿拉伯庚酮糖‑7‑磷酸合成酶的同工酶AroF‑2 内引入了不同的点突变(G136E、G136K 和G136R),其中KT2440::AroF2(G136E)‑PJ23119ΔpcaHΔpykA 菌株摇瓶发酵的儿茶酸产量较AroF‑2 未编辑菌株和原始菌株分别增加了69.01%和611.17%。
中国科学院天津工业生物技术研究所马延和研究团队[128]开发了基于碱基编辑器的一种多重自动化谷氨酸棒杆菌碱基编辑方法(multiplex automatedCorynebacterium glutamicumbase editing method,MACBETH),并利用MACBETH 产生多重基因失活、用于提高谷氨酸产量的突变株文库,发现丙酮酸激酶编码基因pyk和乳酸脱氢酶编码基因ldhA双基因失活菌株可将谷氨酸产量相较野生型提高3 倍。此外,他们通过集成机器人系统实现了MACBETH 的自动化,使该方法能够每月生成数千个定向设计的C. glutamicum代谢工程的菌株。该研究团队利用自动化平台构建了一个包含94 个转录因子基因失活的文库,成功率达到100%,该工作展现了碱基编辑器快速便捷地生成突变菌株库的能力,成为工业应用中多重自动化细菌基因组编辑的强大工具[128]。随后该研究团队利用拓展了PAM序列的Cas蛋白变体(nVQR‑Cas9(D10A)、nVRER‑Cas9 (D10A)、 nxCas9 3.7 (D10A) 和nCas9‑NG(D10A)),替换了MACBETH方法中构建的CBE 中的野生型nCas9,拓展了CBE 在C.glutamicum中的编辑范围,利用nxCas9 3.7(D10A)‑AID去除了lysC基因编码的天冬氨酸激酶产生的反馈抑制,使工程菌株产生1.7 g/L 赖氨酸,而在野生型菌株中,赖氨酸的产量低至几乎无法检测[82]。之后,该研究团队还开发了一种基于碱基编辑器靶向且无模板的表达调控方法(base editor‑targeted and template‑free expression regulation, BETTER)用于在微生物中大规模微调基因的表达,它可以在原位生成大量不同的核糖体结合位点、5'非翻译区或启动子的遗传组合,并无需合成、转化和整合DNA 供体。他们应用BETTER 在模式微生物C.glutamicum和Bacillus subtilis中同时调节多达十个基因的表达,以增强宿主的底物摄取或天然产物生物合成,分别获得改善了木糖分解代谢、甘油分解代谢或番茄红素生物合成的工程菌株[129]。
江南大学周哲敏研究团队[81]构建了可以实现体内原位蛋白进化的工具CRISPR‑CDA‑nCas9‑UGI,并利用该工具在芽孢杆菌B. subtilis中对影响细胞功能的Sec转位酶进行进化突变,获得了提升运输能力的突变株。此外,他们还利用CRISPR‑CDA‑nCas9‑UGI 对抗菌肽抗性决定因子bceB基因进行进化,获得了对抗菌肽抗性更高的突变株。BceB 蛋白在保护细胞壁生物合成靶点免受抗菌肽的抑制方面发挥重要作用,杆菌肽抗性突变体的创建对于构建产生杆菌肽的细胞工厂具有重要价值[81]。
此外,脱氨酶还可以与随机结合DNA 的蛋白融合,设计成为产生DNA 随机突变的工具。浙江大学连佳长研究团队[130]将不同的非特异性ssDNA 结合蛋白与胞苷脱氨酶融合,理论上可以实现在酵母菌Saccharomyces cerevisiae全基因组将胞嘧啶替换成胸腺嘧啶的突变。他们利用这些rBE,提高了S. cerevisiae对异丁醇和乙酸盐的耐受性并增加了产β‑胡萝卜素S. cerevisiae的β‑胡萝卜素产量。他们还使用rBE 对S. cerevisiae基因组进行持续进化,获得了对浓度为9%的异丁醇具有抗性的S. cerevisiae细胞。张学礼研究团队[131]将DNA 解旋酶与活化诱导的胞苷脱氨酶(AID)融合形成酶复合物Helicase‑AID,Helicase‑AID 可以在细胞内整个染色体中随机引入碱基突变。该研究团队利用这一工具分别对产β‑胡萝卜素的E. coliKH001 和S. cerevisiae4742crt 进行多轮随机突变,使得突变株β‑胡萝卜素产量分别增加371.4%和75.4%。
3 展 望
碱基编辑器自问世以来,从最初可以实现目标位点编辑窗口内C 到T、A 到G 的碱基转换,到之后开发和优化出的碱基变换种类不断丰富,编辑窗口、靶向位点不断拓展以及特异性不断优化的新版本,极大地加速了碱基编辑器在生物学、医学等领域的应用。然而,碱基编辑器的碱基编辑偏好性,可以靶向编辑的位点和碱基受限,以及脱靶编辑的问题仍有待进一步完善。例如,虽然目前较多的碱基编辑器在编辑窗口上都比早期版本有所拓展,但是碱基编辑器对于单个或少数碱基的精确编辑仍较难实现,而精确地在基因元件中进行碱基转换对于改造微生物来说也至关重要。此外,碱基编辑器的脱靶编辑所造成的细菌毒性以及微生物的表型变化也会给微生物代谢途径改造带来障碍,虽然目前已报道了一些脱靶效应低至与自发突变水平相当的碱基编辑器,但是这些系统往往包含较多的元件,系统繁复,效能有限,因此开发或者寻找到适合微生物的简便精准的碱基编辑器还需要合成生物学研究工作者的不断努力。同时,还可以通过对以碱基为底物的酶的发现和改造或是对生物体内核酸参与的生化反应的深入了解,利用碱基结构的变化或是体内不同的核酸修复途径,创造更多碱基变换的可能,例如由嘌呤变嘧啶等,将更能拓展碱基编辑器的应用场景。
此外,与其他在微生物中使用的基因编辑工具一样,碱基编辑器也存在着对难以建立遗传操作的微生物无法递送外源DNA 的障碍。而由碱基编辑器实现的碱基转变也需要考虑到微生物产生回复突变的可能性。
碱基编辑器在微生物合成生物学中的应用虽然较CRISPR/Cas 系统的基因编辑有较大优势,但是主要的问题仍旧是碱基编辑所能实现的碱基变换种类较少,虽然可以实现引入提前终止密码子等,但是对于某些氨基酸密码子的转变仍无能为力。因此创造碱基编辑器更多碱基变换的可能变得尤为重要。
可以相信随着人们研究的不断深入,碱基编辑器的性能也将日趋完善,在基础科学、农业和医学等领域将发挥更大的作用。
猜你喜欢
中国实用医药(2021年20期)2021-08-04 14:27:22
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
党的生活·党员电教与远程教育(2019年1期)2019-03-06 12:41:42
铁道通信信号(2018年7期)2018-08-29 01:17:08
中国医药指南(2018年3期)2018-03-23 09:14:25
电子设计工程(2015年4期)2015-02-27 12:04:14
食品工业科技(2012年1期)2012-11-15 02:03:32
