
酸性偏移结合热诱导对大豆分离蛋白微凝胶结构及其特性的影响
2023-09-13 02:52徐彩红王丽娟李佳美孟宪军
食品科学 2023年16期
杨 平,徐彩红,徐 昕,王丽娟,朱 旺,李佳美,孟宪军,许 青,
(1.沈阳师范大学学前与初等教育学院,辽宁 沈阳 110034;2.沈阳师范大学粮食学院,辽宁 沈阳 110034;3.沈阳师范大学实验技术中心,辽宁 沈阳 110034;4.沈阳农业大学食品学院,辽宁 沈阳 110866)
微凝胶是尺寸在微米级及以下,由天然生物大分子构成的多孔三维网状结构,具有高比表面积、多孔、易变形等优点,在食品乳化、功能性物质荷载与递送等方面具有广泛应用前景,是近年来食品科学研究的热点[1]。
蛋白质作为两亲性分子,具有高度的聚集性和良好的胶凝性[2-3],在一定的酸和热诱导下,可以自组装成多种形貌的凝胶结构,如牛奶中的酪蛋白和乳清蛋白、蛋清蛋白、胶原蛋白等均可自组装成微凝胶[4]。大豆分离蛋白(soy protein isolate,SPI)作为天然大分子材料,来源广泛、价格低廉,疏水性氨基酸含量较高,极性和带电残基比例大,是制备食品微凝胶的理想原料。
自组装是基础粒子由无序到有序转变的过程,是一种制备具有吸引特性新粒子的新技术[5]。蛋白质的自组装是分子内和分子间吸引力/排斥力的平衡所控制,自主装作用力的大小不仅与蛋白质自身组成、结构有关,还与其所存在环境密切相关[6]。酸性偏移时蛋白质处于变性与未变性之间的熔球态,其氨基酸残基具有较高的净电荷[7],电荷间的排斥力促使蛋白质结构展开,微环境的变化使其经历去折叠、亚基解离以及再聚集过程,重新自组装成微米颗粒[8],Jiang Jiang等[9]研究发现酸性偏移可以改变大豆蛋白结构,提高其乳化性;李次力等[10]发现酸性偏移使大豆亲脂蛋白发生自组装;热诱导能提供的额外分子间斥力,改变蛋白质暴露出的官能团,提高蛋白质颗粒的表面疏水性,改变蛋白质的结构[11]和功能[12],乳清蛋白经过高温可形成稳定性高、耐热性好的球形网状微凝胶[13],王健等[14]发现酸性偏移能增加SPI在加热过程中的柔性,使其结构更加舒展。蛋白质变性程度不同,会导致其表面氨基酸组成、电荷分布的差异,进而形成不同的微凝胶结构,不同蛋白质改性方法相结合,可以更加精准的定向调控蛋白质变性程度。因此,本研究通过酸性偏移结合热诱导对蛋白质进行胶凝化改性,探究热诱导温度对微凝胶结构及其特性的影响,精准调控蛋白质的自组装,为合理开发植物蛋白微凝胶提供理论支持和实践依据。
1 材料与方法
1.1 材料与试剂
脱脂豆粕(纯度94.2%)山东禹王实业有限公司;硫磺素T(thioflavin-T,ThT)、8-苯氨基萘-1-磺酸(8-phenylaminonaphthalene-1-sulfonic acid,ANS)美国Sigma-Aldrich公司;其他试剂均为国产分析纯。
1.2 仪器与设备
F97 Pro荧光分光光度计 上海棱光技术有限公司;VEC-TOR33傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)仪、Dimension Icon原子力显微镜 德国Bruker公司;DSC 200 F3差示扫描量热仪德国耐驰公司;S3500激光粒度分析仪 美国Microtrac公司;FE28 pH计 梅特勒-托利多国际贸易(上海)有限公司;Scientz-12N冷冻干燥机 宁波新芝生物科技股份有限公司;X-12R高速冷冻离心机 美国Beckman公司;3150紫外-可见分光光度计 日本岛津公司;DF-101S恒温加热磁力搅拌器 上海力辰邦西仪器有限公司。
1.3 方法
1.3.1 SPI的提取
采用碱溶酸沉方法提取SPI,用10 倍质量的去离子水溶解低温脱脂豆粕粉,调节pH值至8.0、室温(25 ℃)搅拌2 h后,8000 r/min离心20 min,取上清液。再调节pH值至4.6,4 ℃静置1 h后,上述条件再次离心,用2 mol/L NaOH溶液搅拌至沉淀充分溶解,调节pH值至7.0,4 ℃透析48 h,冷冻干燥后备用。
1.3.2 大豆分离蛋白微凝胶(soy protein isolate microgel,SPIM)制备
参考Li Ting等[15]方法稍作修改。用超纯水配制10 mg/mL的SPI溶液,4 ℃水化过夜,3 mol/L HCl溶液调节pH值至2.0,15000 r/min离心15 min,取上清液。经6~8 kDa透析袋透析24 h后,稀释至2 mg/mL,在不同温度(25、45、55、65、75、85 ℃)条件下,以200 r/min恒温搅拌12 h后4 ℃冷却,得到SPIM初始液,冷冻干燥后保存。不同温度下制备的微凝胶,分别记作25SPIM、45SPIM、55SPIM、65SPIM、75SPIM、85SPIM。
1.3.3 ThT荧光光谱分析
参考Gharanjig等[16]方法稍加修改。取质量浓度为2 mg/mL SPIM样品溶液40 μL,经过0.24 μm滤膜过滤,与4 mL ThT工作液(0.4 mg/mL)充分混合后,进行荧光光谱分析。设定激发波长为440 nm,发射波长为488 nm,激发狭缝和发射狭缝为5 nm。
1.3.4 二级结构测定
取1~2 mg冻干的样品与200 mg KBr充分研磨混匀,150 MPa加压5 min后,FTIR扫描样品[17],扫描范围为4000~400 cm-1,分辨率为4 cm-1。PeakFit 4.12软件分析酰胺I带(1600~1700 cm-1),用峰面积比率表示蛋白质二级结构的相对含量。
1.3.5 表面疏水性分析
采用ANS法[18]进行分析。取4 mL质量浓度为1 mg/mL的样品与20 μL的8 mmol/L ANS充分混合,避光反应10 min后进行荧光分析,设定激发波长为365 nm,发射波长为484 nm。
1.3.6 粒度测定
取4 mL质量浓度为1 mg/mL的样品,经0.45 μm滤膜过滤后,采用激光粒度分析仪测定样品粒度,将样品累计粒度分布达到10%、50%、90%时的粒度值,分别记作Dx(10)、Dx(50)、Dx(90)。
1.3.7 差示扫描量热分析
称取50 μL质量浓度为20 mg/mL的样品,置于密封坩埚中,设定加热温度为20~200 ℃,升温速率为10 ℃/min,利用差示扫描量热仪自带的软件,计算变性温度(Tm)和焓变(ΔH)。
1.3.8 微观结构观察
采用原子力显微镜轻敲模式观察样品的微观形貌,设定频率为320 kHz,扫描速率为1 Hz,silicon tips长度为125 μm,曲率半径为42 N/m。
1.3.9 乳化性分析
参考栗俊广等[19]方法,并作适当修改。取50 μL质量浓度为10 mg/mL的SPIM样品与5 mL质量分数0.1%的十二烷基硫酸钠溶液充分混匀,用紫外分光光度计于500 nm波长处测定吸光度。按照下式计算乳化活性指数(emulsifying activity index,EAI)和乳化稳定性指数(emulsion stability index,ESI):
式中:ρ为SPIM质量浓度/(g/mL);φ为油相体积分数(20%);A0和A10为静置时间为0 min和10 min的吸光度。
1.3.10 持水性的测定
准确称取2 g的SPIM于离心管中,4 ℃、8000 r/min离心15 min后,称量沉淀的质量,按下式计算凝胶持水率:
式中:m1为离心前SPIM的质量/g;m2为离心后SPIM的质量/g。
1.4 数据分析
采用SPSS 21.0软件进行数据处理,单因素方差分析,实验数据用表示,P<0.05,差异显著,有统计学意义,并采用Origin 2019软件作图。
2 结果与分析
2.1 ThT荧光分析结果
ThT可以与蛋白质中β-折叠结构特异结合,β-折叠相对含量越多则ThT荧光强度越强[20]。图1显示,与25SPIM相比,酸性偏移结合热诱导后SPIM的荧光强度增加,其中75SPIM和85SPIM的荧光强度最大,这可能由于酸性偏移环境使SPI表面呈现大量正电荷,分子间的静电斥力增加,而酸性偏移结合热诱导后,蛋白质体积发生一定程度的膨胀,使其表面静电荷密度相对降低,分子间静电斥力减小,部分结构互相靠近形成β-折叠结构[21]。
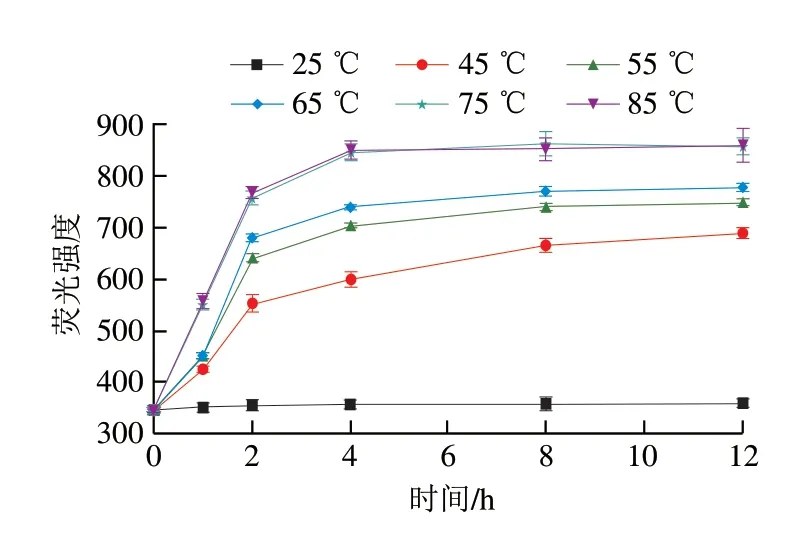
图1 SPIM的形成动力学曲线Fig.1 Kinetic curves of SPIM formation
从图1还可以看出,SPIM形成动力学曲线的最初2 h内荧光强度增长速率最快,而后速率逐渐减缓,在4~12 h处荧光强度达到最高并保持平稳。SPIM形成初期(0~4 h),样品ThT荧光强度增大,可能是由于酸性偏移结合热诱导后蛋白质二级结构发生转变[22],呈现“折叠-去折叠-复折叠”的变化,β-折叠相对含量增加[23]。平衡阶段(4~12 h),样品ThT荧光强度保持平稳,表明蛋白质二级结构转变基本完成,β-折叠相对含量稳定,此阶段以β-折叠作为自组装的“构建单元”,相互交联形成微凝胶[15]。
2.2 二级结构分析结果
FTIR可以分析不同热诱导温度对蛋白质二级结构的影响,以及微凝胶形成前后结构的变化。如图2所示,25SPIM的酰胺I带特征峰在1635 cm-1,酰胺II带在1535 cm-1,而微凝胶形成后酰胺I带的特征峰红移至1610 cm-1附近,酰胺II带的特征峰红移至1525 cm-1附近,酰胺A带(3000~3600 cm-1)吸收峰的强度和位置也发生改变,这表明静电作用、疏水作用、氢键作用[24]参与了SPIM的自组装。
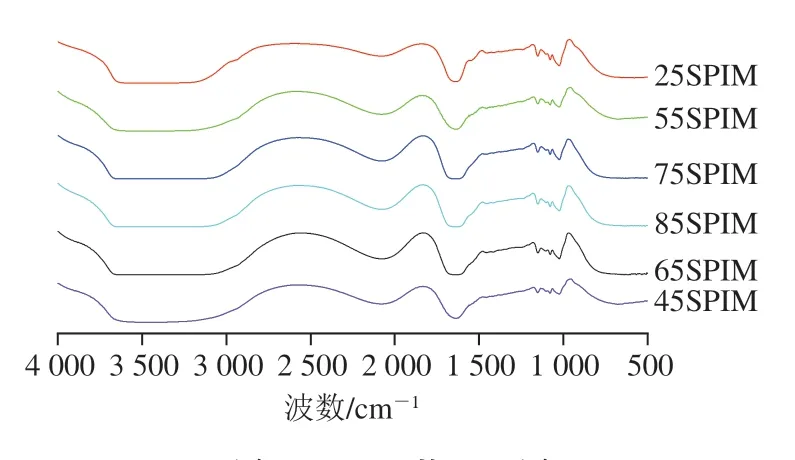
图2 SPIM的FTIR图Fig.2 Fourier transform infrared spectra of SPIM
二级结构是蛋白质发挥功能的基础,当α-螺旋/β-折叠比值降低时蛋白质的有序性会增加,结构变得疏松而有序、柔性增加[25],当β-折叠相对含量增加、β-转角相对含量降低时,蛋白质的稳定性会增加[26]。通过PeakFit软件去卷积拟合FTIR的酰胺I带,可以定量评价蛋白质二级结构的分类和比例,其中β-折叠、无规卷曲、α-螺旋、β-转角结构对应波数分别为1610~1639、1640~1649、1650~1659、1660~1700 cm-1[27]。去卷积拟合后得到的各类二级结构占比(图3),可见随着温度升高β-转角相对含量逐渐减少,β-折叠相对含量逐渐增加,α-螺旋/β-折叠比值逐渐降低,其中75SPIM和85SPIM组的比值最小,均约为0.23,比25SPIM组降低了48.89%,这表明酸性偏移结合热诱导增加了SPIM的柔性和稳定性。
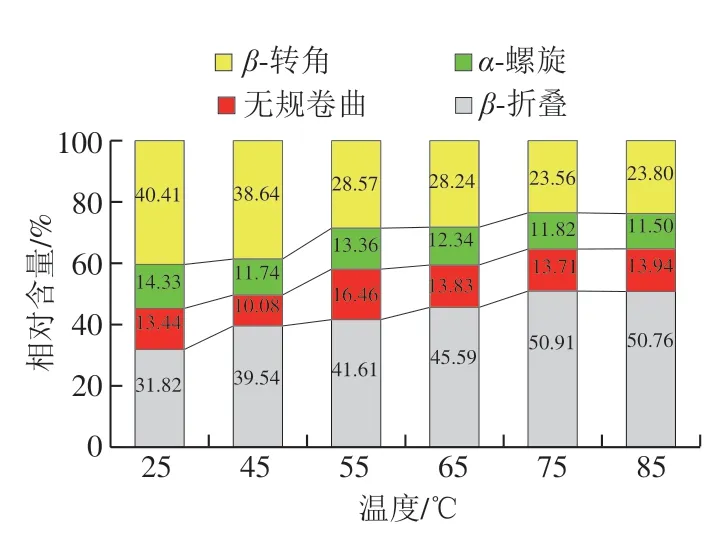
图3 SPIM的二级结构组成Fig.3 Secondary structure composition of SPIM
2.3 表面疏水性分析结果
疏水相互作用是维持蛋白质三级结构的主要作用力,表面疏水性指数可以表征蛋白质分子表面疏水基团的相对含量,是评价蛋白质构象变化的重要参数[28]。图4显示,酸性偏移结合热诱导后,SPIM表面疏水性指数随着温度升高呈现先上升后下降的趋势,其中65SPIM表面疏水性指数最大,显著高于其他各组(P<0.05),比25SPIM增加了43.98%。这可能是由于酸性偏移结合热诱导会加速蛋白质高级结构解折叠,使其内部疏水性基团暴露,ANS荧光探针结合位点增多[29];但当热诱导温度大于65 ℃时,大量疏水基团暴露于蛋白分子表面,疏水效应使疏水基团彼此靠近,蛋白质分子重新聚集,表面疏水性指数降低。
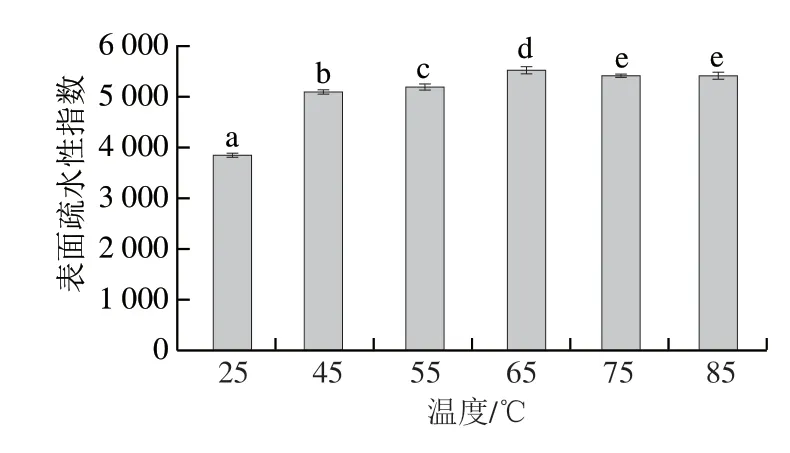
图4 SPIM的表面疏水性Fig.4 Surface hydrophobicity of SPIM
2.4 粒度分析结果
粒度分析可以阐明蛋白质聚集行为对其凝胶特性的影响。酸性偏移结合热诱导后样品的粒度分析结果如图5、表1所示,各组样品的粒度均在1000 nm以下,属于微凝胶颗粒。如图5所示,25SPIM粒度呈三峰分布,与其相比,45SPIM粒度分布3 个峰均左移,粒度变小;当热诱导温度大于55 ℃时,SPIM粒度分布转为单峰,峰形态趋近于正态分布,表明酸性偏移结合热诱导改变了蛋白质的形态。表1显示,当热诱导温度大于55 ℃时,随着温度的升高Dx(50)值逐渐增加,这可能是由于自组装是热力学平衡条件下进行的分子重排过程,热诱导使自组装的“构建单元”运动加剧,有效碰撞机率增加,导致了平均粒度的增加。表1还显示,随着热诱导温度升高,比表面积逐渐增加,其中75SPIM的比表面积最大,为25SPIM的9.26 倍,显著高于其他各组(P<0.05),但当温度大于75 ℃时,SPIM比表面积降低,表明热诱导温度过高不利于其比表面积的继续增加。
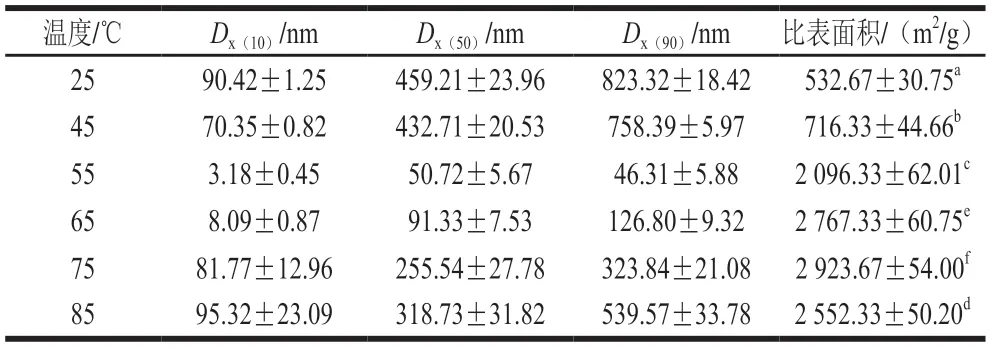
表1 SPIM的粒度和比表面积Table 1 Particle size and specific surface area of SPIM
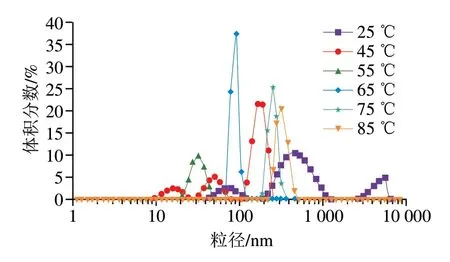
图5 SPIM的粒度分布Fig.5 Particle size distribution of SPIM
2.5 差示扫描量热分析结果
差示扫描量热法可以分析热诱导引起的蛋白质构象变化。Tm值为热变性过程中,蛋白质中折叠状态和去折叠状态的含量相等时的温度,其值越大蛋白质越稳定;ΔH表示蛋白质变性需要的能量,ΔH增大则需要更多的能量使蛋白质去折叠,蛋白质结构更紧凑[30]。表2显示,酸性偏移结合热诱导后,随温度升高样品的Tm和ΔH值呈现先降低后升高的趋势,其中55SPIM的Tm和ΔH值最低,75SPIM和85SPIM最高。这可能是由于酸性偏移结合低温热诱导引起了蛋白质结构解折叠,热稳定性较低,而随着热诱导温度继续升高,蛋白质自组装成结构紧凑的微凝胶,热稳定性增加。

表2 SPIM的变性温度和焓变值Table 2 Denaturation temperature and enthalpy change of SPIM
2.6 微观形态观察结果
图6显示酸性偏移结合不同温度热诱导后,SPIM微观形态发生了明显变化。25SPIM呈现球状结构,颗粒大小均匀;45SPIM球状结构部分消失,颗粒大小不均匀,出现细小的颗粒样结构;55SPIM呈现长度较短的棒状结构;65SPIM呈现树形聚集物,分枝细长;75SPIM和85SPIM凝胶结构相似,都具有网状的微观结构。这可能由于酸性偏移结合低温热诱导时,自组装的“构建单元”浓度较低,其端基相连成棒状结构[31],随着热诱导温度升高,“构建单元”浓度增加,聚集成为空间网状结构。由图6还可见,75SPIM的孔径较均匀且规则,网状结构连续,而85SPIM部分网状结构连续部分中断,孔径大小不一,出现热诱导过度现象。


图6 SPIM的微观形态Fig.6 Microscopic morphology of SPIM
2.7 EAI及ESI分析结果
如图7所示,随着热诱导温度的升高,样品的EAI和ESI呈现先升高后下降的趋势。75SPIM的EAI及ESI显著高于其他各组(P<0.05),分别为87.17 m2/g和65.55%,较25SPIM分别提高了30.87%和85.54%,这可能由于SPIM二级结构中α-螺旋/β-折叠比值降低,凝胶柔性升高,利于蛋白-界面相互作用,可以承受更高的外界干扰,乳液的稳定性提高[32]。与75SPIM相比,85SPIM的EAI及ESI显著降低(P<0.05),这可能与其微观结构的孔径大小不均匀、网状结构不连续、比表面积减小、吸附能力下降有关。
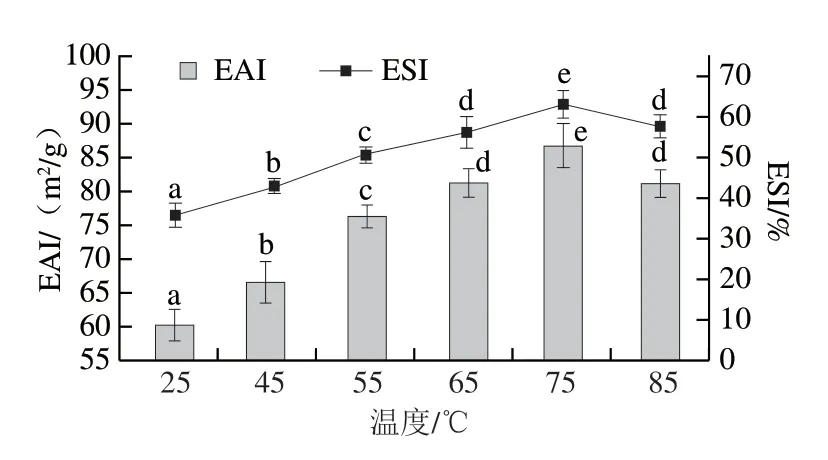
图7 SPIM的EAI和ESIFig.7 EAI and ESI of SPIM
2.8 持水性分析结果
微凝胶的持水性可以反映其空间网状结构的紧密程度。图8显示,随着热诱导温度升高,微凝胶持水性呈现阶段性变化,其中25~55 ℃阶段,随着温度的升高,持水率呈现增加的趋势,即55SPIM持水性最高,这表明热诱导增加了微凝胶吸附和结合水的能力,且具有较强的稳定性和抗干扰能力。55~85 ℃阶段,持水性呈现降低的趋势,这可能与其微观结构较紧密,基质中存在未结合的过量水,离心后这些水被释放出来有关[33]。本研究持水率的变化与蛋白质二级结构中α-螺旋相对含量变化趋势一致。
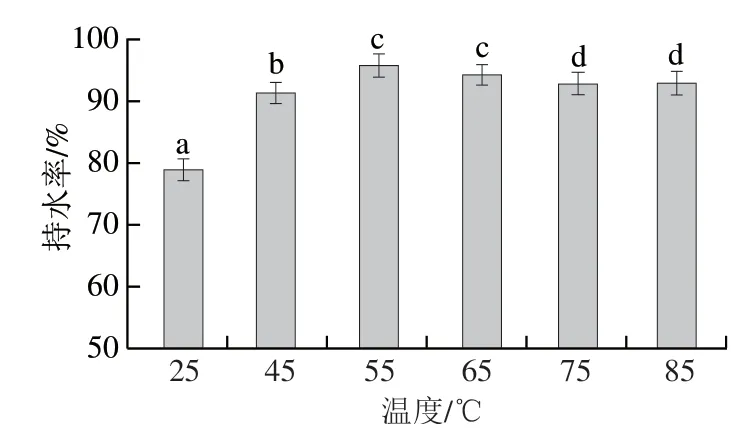
图8 SPIM的持水性Fig.8 Water-holding capacity of SPIM
3 结论
通过酸性偏移结合热诱导对SPI进行胶凝化改性,对比热诱导温度对SPIM结构及其凝胶特性的影响,发现适度的热诱导能使SPI自组装成孔径大小均匀的网状微凝胶。结果表明,酸性偏移结合热诱导提高了蛋白质中β-折叠相对含量,静电相互作用、疏水相互作用、氢键作用参与了微凝胶的自组装。随着热诱导温度升高,SPIM的表面疏水性指数先上升后下降,热稳定性逐渐增强。与25SPIM相比,75SPIM的比表面积增加,乳化活性、乳化稳定性增强,持水性升高。综上所述,酸性偏移结合热诱导是一种调控蛋白质微凝胶结构和特性的有效方法,通过精准控温可以提升微凝胶的质量。
猜你喜欢
云南化工(2021年10期)2021-12-21
粉末冶金技术(2021年3期)2021-07-28
南京大学学报(自然科学版)(2021年1期)2021-01-30
福建基础教育研究(2019年8期)2019-05-28
意林·全彩Color(2018年9期)2018-10-12
中学生数理化·八年级物理人教版(2017年6期)2017-11-09
系统工程与电子技术(2016年12期)2016-12-24
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
橡胶工业(2015年2期)2015-07-29
河北大学学报(自然科学版)(2015年1期)2015-02-27
