
Network Pharmacology, Molecular Docking and Experimental Exploring Molecular Mechanisms of Yinqiao Anti-Epidemic Formula in Regulating Mucosal Immune System of Respiratory Tract
2023-09-07 14:33:58HengXU,FangyuanLI,GuoqiangLIANG,JieYUAN1,XiudaoSONG,YunhaiZHOU,HuiZHU,YichaoYAN,TingtingWU
农业生物技术(英文版) 2023年4期
Heng XU, Fangyuan LI, Guoqiang LIANG, Jie YUAN1, Xiudao SONG, Yunhai ZHOU, Hui ZHU, Yichao YAN, Tingting WU

Abstract[Objectives] To explore the molecular mechanisms of Yinqiao anti-epidemic formula (YQAEF) in regulating mucosal immune system of respiratory tract. [Methods] The active components of YQAEF were obtained from the TCMSP database, and RMIS targets were obtained from the GeneCards database. A "YQAEF components-RMIS targets-pathways" network was constructed by analyzing the above data to screen core targets for molecular docking verification. A mouse model of acute upper respiratory tract infection (AURI) was developed. Based on the experimental models, the key pathway target genes screened by network pharmacology were verified in vivo. [Results] The main active components of YQAEF involved in the regulation of the RMIS included quercetin, acetic acid, and raffinose. Key targets, such as angiotensin-converting enzyme (ACE), galactosidase alpha (GLA), matrix metalloproteinase 2 (MMP2), Serpin Family E Member 1 (SERPINE1), and myeloperoxidase (MPO) and important viral infection and endocrine resistance signaling pathways were included in the regulation of the RMIS with YQAEF. Molecular docking assays showed that the key targets had good binding activities with the components of YQAEF. Based on the results of network pharmacology, key target proteins in ACE, GLA, MMP2, SERPINE1, and MPO were selected for experimental verification. The results showed that ACE/ACE2 and MPO expressions were increased in the oral and throat mucosa of the AURI mice. Under YQAEF treatment, the expression levels of ACE/ACE2 and MPO were decreased. [Conclusions] This study revealed the mechanism of YQAEF in the regulation of RMIS, which is associated with multiple components, targets, and pathways. Further experiments confirmed that YQAEF interfered with MPO and ACE/ACE2 signaling pathways to regulate the RMIS in the oral and throat mucosa tissue of mice with AURI, and provide a new direction for exploring the potential antiviral mechanism of YQAEF.
Key wordsRespiratory passage mucosal immune system; RMIS; Network pharmacology; Molecular docking; ACE/ACE2, Yinqiao anti-epidemic formula (YQAEF)
DOI:10.19759/j.cnki.2164-4993.2023.04.017
Since the end of 2019, there has been a global outbreak of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2, formerly known as 2019-nCoV). The spread of SARS-CoV-2 has not been fundamentally suppressed[1]. The world attention is focused on the traceability, transmission route, and pathogenesis of the new coronavirus. There is also a corresponding interest in novel therapeutic drugs and vaccines; production, research and development of medical protective materials; rapid diagnosis techniques; and prevention and control strategies. Integrated traditional Chinese and Western medicine may offer hitherto unexplored avenues of investigation[2].
The current research on the prevention and treatment of COVID-19 from the perspective of mucosal immunity is prospective and holistic, and better reflects the characteristics of traditional Chinese medicine (TCM). According to TCM principles, the immune function of the body is the main manifestation of righteousness, and it is the main part of the body that fights viral infection. Strengthening the righteousness and eliminating evil spirits of the body has always been the characteristic and advantage of TCM in the treatment of many infectious diseases[3]. By regulating the immune function state of local disorders of the mucosa, TCM improves the immune function of the body and has a positive effect on enhancing the ability of resistance and repair[4]. The respiratory passage mucosal immune system (RMIS) provides a new way to elucidate the mechanism of action of TCM in the treatment of COVID-19, and provides inspiration for the prevention and treatment of COVID-19[5]. The application of special TCM in the treatment of the RMIS for virus infection possesses the advantages of multi-target, multi-level treatment mechanisms, few adverse reactions, high safety, and unique advantages for the prevention and treatment of virus infection, which have been frequently highlighted[6].
Yinqiao anti-epidemic formula (YQAEF) is a Chinese medicine preparation based on the traditional Chinese prescriptions Yinqiao San ("Warm Febrile Diseases") [7], Yupingfeng San ("Yifangleiju"), and Huoxiang Zhengqi powder ("Usage Diversity") [8-9]. It is combines Lonicerae Japonicae Flos, Forsythiae Fructus, Pogostemon Cablin Benth, Hedysarum Multijugum Maxim, Atractylodes Macrocephala Koidz, Sposhnikoviae Radix, Platycodon Grandiforus, PLablab Semen Album, and Licorice. It not only has the functions of heat-clearing, detoxicating, and removing dampness for stomach regulation, but also strengthens the spleen, supplements the lungs, and promotes healthy qi to eliminate pathogens. Attack and supplement, as well as internal and external treatments, are suitable for most peoples constitutions and have many effective clinical applications. Therefore, it exerts an antiviral effect[10].
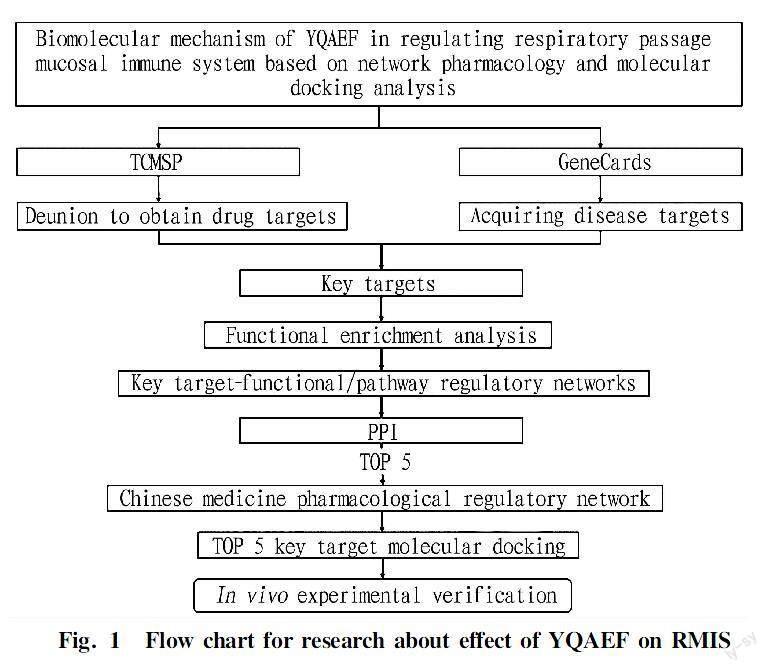
Network pharmacology is based on bioinformatics and computer technology, which integrate a large amount of biological information and data. It systematically and comprehensively reflects the intervention mechanism of drugs on the disease network, which is similar to the mechanism by which the complex components of traditional Chinese medicines interfere with diseases through multiple channels and multiple targets[11]. In this study, we aimed to explore the targets of YQAEF in the regulation of the RMIS and to elucidate the therapeutic mechanism of YQAEF on acute upper respiratory tract infection (AURI) from many aspects. Furthermore, some target proteins of key signaling pathways were verified through in vivo experiments, which may provide evidence for the treatment and prevention of the spread of the virus using YQAEF. A flowchart is shown in Fig. 1[12-13].
Materials and Methods
Components database construction and ADME screening
The candidate chemical components of the nine herbs contained in YQAEF and the putative targets of these components were identified with the TCM Systems Pharmacology (TCMSP, http://lsp.nwu.edu.cn/tcmsp.php, Ver.2.3) database using "Lonicerae Japonicae Flos," "Forsythiae Fructus," "Pogostemon Cablin (Blanco) Benth," "Hedysarum Multijugum Maxim," "Atractylodes Macrocephala Koidz," "Saposhnikoviae Radix," "Platycodon Grandiforus," "PLablab Semen Album," and "Licorice" as the search terms[14]. The candidate active components were screened based on absorption, distribution, metabolism, and excretion properties such as oral bioavailability (OB) and drug similarity (DL) [15]. The filtering criteria used were OB≥30% and DL≥0.18.
Predicting the targets of RMIS
The GeneCards database (https://www.genecards.org/) and the Therapeutic Target Database (TTD) (https://db.idrblab.org/ttd/) were used to gather information on RMIS-associated target genes. GeneCards is a comprehensive database of functions involving proteomics, genomics, and transcriptomics[16]. Target names were collected from TTD, which provided information on the therapeutic protein targets, and the corresponding ID of each target[17].
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis
The drug targets and disease targets were intersected, and GO and KEGG pathway enrichment analysis was performed using the R language and Cluster Profiler (version 3.18.0) software package[18-20]. Statistical significance was set at P<0.01, and Cytoscape (version 3.8.2) was used to visualize and analyze the results[21]. Further, a regulatory network diagram of the intersection target-function and intersection target-pathway was drawn.
Construction of protein-protein interactions (PPI) network
The intersection of the drug target of YQAEF and the target of the RMIS was selected to draw the Venn diagram using Venny (version 2.1.0). The intersection target was entered into the STRING11.0 database (https://string-db.org) to construct a PPI network[13, 22]. Cytoscape was used to construct and visualize the PPI network. CytoNCA, a network topology analysis plug-in in Cytoscape, was used for the topological analysis of PPI networks. "Degree" refers to the number of connections of the node in the whole network, and reflects the interaction information between nodes. The "Degree" value was thus used as a reference for the importance of the core target. The active components corresponding to the five key targets obtained above were further proposed. A herbs-active component-key target gene network was constructed, and Cytoscape was used to visualize and analyze the results.
Screening of key genes and verification by molecular docking
Using the "CytoHubba" plug-in in Cytoscape software, the top five genes in the PPI network were screened out as key genes based on the "Degree" value[23]. The crystal structure of the target protein was downloaded from the PDB database (https://www.rcsb.org/). PyMOL (version 2.5) and AutoDock (version 1.5.6) were used to remove water and the original ligand of the active center, and to hydrogenate the target protein. The 3D structural files of the core components of TCM were downloaded from the PubChem database (https://www.ncbi.nlm.nih.gov/pccompound/). They were then loaded into AutoDock Tools to add atomic charges and assign atomic types. All the flexible bonds were set to be rotatable. AutoDock Vina was used for molecular docking. The conformation with the highest score was selected as the docking conformation, and PyMol was used to visualize the results[12].
Experimental verification
AnimalsSix-week-old female ICR (Institute of Cancer Research) mice (body weight 20-22 g) were routinely housed in SPF animal breeding rooms. Permission was obtained from the Animal Care and Use Ethics Committee of our hospital. All procedures were performed in accordance with the committee guidelines.
Drugs and reagentsThe YQAEF was prepared at the Clinical Pharmaceutical Laboratory of Traditional Chinese Medicine of the Suzhou Academy of Wumen Chinese Medicine. The herbs were boiled in water three times for 1 h each time. The decoctions were combined and filtered through a membrane, then concentrated through vacuum evaporation to a density of 1.0 at 70 ℃. RIPA lysis buffer, BCA Protein Assay Kit, Tris-glycine-SDS transfer buffer, Tris-glycine-SDS electrophoresis buffer, SDS-PAGE protein loading buffer, Western blot transfer filter paper, PVDF membrane, ultra-sensitive ECL chemiluminescence solution, and 4% paraformaldehyde fixative were purchased from Shanghai Beyotime Biotechnology Co., Ltd. Primary antibodies against rabbit immunoglobulin A (IgA), immunoglobulin receptor (pIgR), angiotensin-converting enzyme (ACE), galactosidase alpha (GLA), serpin family E Member 1 (SERPINE1), myeloperoxidase (MPO), angiotensin-converting enzyme 2 (ACE2), and glyceraldehyde-3-phosphate dehydrogenase as well as secondary antibodies were purchased from Abcam.
Animal model and treatmentsAfter 1 week of acclimation to the environment, ICR mice were randomly divided into three groups with 10 mice per group: non-treated control group, model group, and YQAEF (10 g/kg) group. The YQAEF powder was dissolved in deionized water for oral gavage once daily at the doses mentioned above to the YQAEF group alone. Mice in the control and model groups received an equivalent volume of deionized water over the same treatment period. The mice had unlimited access to food and water. The treatment duration was 1 week. Except for the control group, the mice in the other groups replicated the Upper airway immunity dysfunction (UAID) model on the 7th day, which involved exposure to a cold environment at -20 ℃ for 15 min, followed by acclimation at room temperature for 1 h[24]. The mice in each group were euthanized after anesthesia 1 h after the last administration, and their oral and throat mucosa were collected in 1.5-mL EP tubes and stored at -80 ℃.
Detection of target protein by western blot (WB) analysisTotal protein was extracted and collected from the oral cavity and larynx tissue. The protein was denatured using loading buffer and then separated by SDS-PAGE (8%). The protein in the gel was then transferred to a PVDF membrane using a Bio-Rad electro transmitter. Subsequently, 5% skim milk powder was used to seal the PVDF membrane at room temperature. After blocking, the primary antibody was added and incubated overnight at 4 ℃. The next day, the washed PVDF membrane was incubated with horseradish peroxidase-labelled secondary antibodies at room temperature. After 1 h of incubation, the strips on the PVDF membrane were developed in a dark room with ECL chemiluminescence reagent, and each WB analysis was performed three times. Semi-quantitative analysis was performed using ImageJ software.
Detection of target proteins using immunohistochemistry (IHC)Sections (3 μm) of paraffin-embedded oral cavity and larynx tissues were deparaffinized and rehydrated, followed by incubation with 3% H2O2 for 10 min to suppress the endogenous peroxidase activity. The sections were then incubated overnight at 4 ℃ with diluted primary antibodies against ACE and ACE2. The slides were incubated with biotin labeled with goat anti-rabbit IgG (secondary antibody) at 37 ℃ for 15 min. They were then orderly incubated with DAB reagent and hematoxylin (for 2 min). The slides were then dehydrated, made transparent, and mounted with neutral gum. The tissue sections were examined under an optical microscope and quantitative analysis was performed using Image-Pro Plus 6.0 medical pathology graphic color analysis software.
Heng XU et al. Network Pharmacology, Molecular Docking and Experimental Exploring Molecular Mechanisms of Yinqiao Anti-Epidemic Formula in Regulating Mucosal Immune System of Respiratory TractStatistical Analysis
Data 20.0 was used for statistical analysis, and measurement data were expressed as mean ± standard deviation (x±s). One-way analysis of variance was used for comparison between multiple groups. For comparison, the LSD test was used when the variance was homogeneous and the non-parametric test was used for the heterogeneity of variance. P<0.05 represents a statistically significant difference, while P<0.01 and P<0.001 represent a statistically significant difference.
Results and Analysis
Construction of YQAEF active component-target network
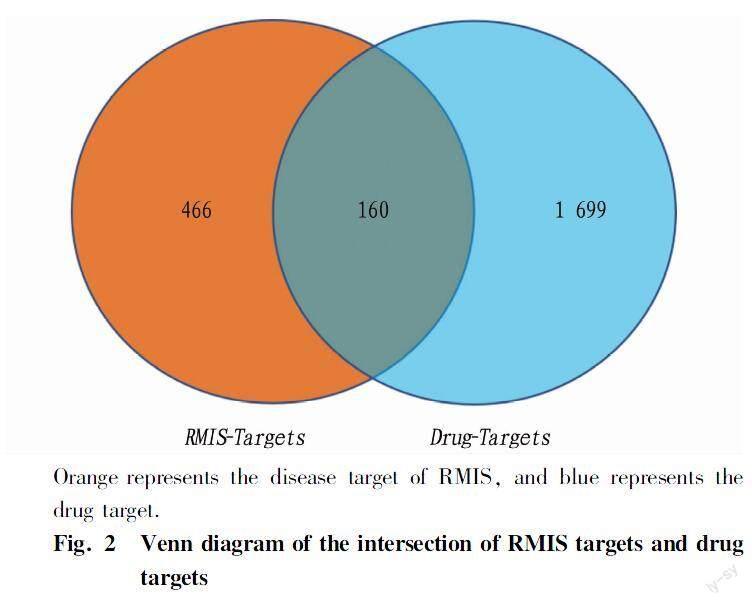
The targets corresponding to each drug-active compound were screened using the TCMSP database. The active compounds primarily originated from Lonicerae Japonicae Flos (211 compounds), Forsythiae Fructus (206 compounds), Pogostemon Cablin Benth (174 compounds), Hedysarum Multijugum Maxim (181 compounds), Atractylodes Macrocephala Koidz (225 compounds), Saposhnikoviae Radix (520 compounds), Platycodon Grandiforus (84 compounds), PLablab Semen Album (60 compounds), and licorice (157 compounds). In this study, the drug targets obtained for each disease were merged, and finally 638 active components, 626 drug targets, and 1 859 related disease targets were obtained, and the R language VennDiagram package was used to analyze the above obtained drug targets and RMIS diseases. The targets were intersected and 160 key targets were obtained (Fig. 2).
GO pathway enrichment analysis
To further understand the intersecting genes, GO enrichment analysis was performed. This project used the R software clusterProfiler package to conduct enrichment analysis based on the GO database to find the common functions and related pathways of a large number of genes in the key gene set of the module. The screening conditions were P<0.05, and counts>2. The GO system includes three parts: biological processes (BP), cellular components (CC), and molecular functions (MF). In this study, GO functional annotation was performed on 16 key target genes, and the biological significance of each gene was determined.
In this study, the R language "enrichplot" (version 1.10.2) was used to draw bar graphs to display the GO functional enrichment results. The results showed that in terms of BP, a total of 1770 terms were obtained, and key target genes were significantly related to drug response, oxidative stress response, and inflammatory response, including regulation of inflammatory response, positive regulation of establishment of protein localization, positive regulation of protein transport, reactive oxygen species metabolic process, cellular response to oxidative stress, response to drug, response to oxidative stress, response to molecule of bacterial origin, response to lipopolysaccharide, and cellular response to chemical stress. In terms of MF, a total of 121 terms were obtained, and key target genes were significantly related to organic acid binding, heme binding, and antioxidant activity, including serine-type peptidase activity, amide binding, antioxidant activity, serine hydrolase activity, organic acid binding, tetrapyrrole binding, carboxylic acid binding, heme binding, ligand-activated, transcription factor activity, and nuclear receptor activity. In terms of CC, a total of 81 terms were obtained, and the key target genes were significantly related to the membrane region, outer side of the plasma membrane, and secretory granule cavity, including ficolin-1-rich granules, external side of plasma membrane, caveola, plasma membrane raft, secretory granule lumen, cytoplasmic vesicle lumen, vesicle lumen, membrane region, membrane microdomain, and membrane raft (Fig. 3A-B).
We used the top 10 terms of GO-BP, GO-CC, GO-MF results (the terms GO-BP, GO-CC, and GO-MF cannot be fully displayed) and their corresponding key targets were proposed. Using Cytoscape, the key target-function regulatory network diagram was constructed and visualized, as shown in Fig. 4A. The key target-function network diagram of the GO-BP top 10 terms contains 10 terms, 93 key targets, and 315 relationship pairs. Fig. 4B presents the key target-function network diagram of GO-MF terms. The Figure contains 10 terms, 64 key targets, and 131 relationship pairs. Fig. 4C presents the key target-function network diagram of the GO-CC terms, which contains 10 terms, 59 key targets, and 194 relationship pairs.
KEGG pathway enrichment analysis
The KEGG database is commonly used for pathway research. KEGG pathway analysis was performed on all identified protein sets or screened differentially expressed proteins to perform pathway annotation and to analyze the most important metabolic and signal transduction pathways involved in these proteins or genes. We performed KEGG functional enrichment analysis on 16 key target genes, obtained 176 related pathways, and used the R language "enrichplot" package to draw a bubble chart to display the top 15 pathways. As shown in (Fig. 5A-B), KEGG enrichment results include pertussis, TNF signaling pathway, endocrine resistance, prostate cancer, IL-17 signaling pathway, toxoplasmosis, Chagas disease, tuberculosis, hepatitis B, AGE-RAGE signaling pathway in diabetic complications, Kaposi sarcoma-associated, herpesvirus infection, chemical carcinogenesis-receptor activation, fluid shear stress and atherosclerosis, PI3K-Akt signaling pathway, and lipid and atherosclerosis pathways. This indicates that the key target genes are significantly related to signaling, viral infection, and endocrine resistance.
We identified the KEGG top 50 pathways and their corresponding key targets, constructed a regulatory network diagram of the key target pathways, and used Cytoscape for visualization (Fig. 5C). The network contained 50 KEGG Pathways and 100 key targets, with a total of 891 relational pairs.
Construction of PPI network
The protein interaction network uses proteins as nodes and participates in the same metabolic pathway, biological process, structural complex, functional association, or physical contact between proteins as an edge network. Protein interaction networks are currently among the most well-studied biomolecular networks. Proteins are important biological macromolecules that are found in organisms and perform various biological functions. Proteins participate in all aspects of life processes, such as biological signal transmission, gene expression regulation, energy and material metabolism, and cell cycle regulation, through interaction to form a network. Therefore, protein interaction networks are crucial for understanding the structure and function of cellular networks, and the basis of disease development[25].
First, the relevance score (obtained from the GeneCards database) of 160 key targets was ranked, and 80 genes (Table 1) with a relevance score greater than or equal to the median (median=18.124 248 5) were used for subsequent analysis. To explore whether there was an interaction between key targets, we used the STRING website to construct a PPI network for 80 key targets (confidence=0.4). Discrete proteins were removed to obtain the interaction network relationship of 80 proteins, including 80 nodes and 2 510 edges. Cytoscape software was used to identify the protein network map (Fig. 6). The degrees of the genes in the PPI network were then sorted, and the TOP 5 genes were screened by degree (Table 2) for subsequent analysis. It can be seen that ACE, GLA, MMP2, SERPINE1, and MPO may play an important role in the occurrence and development of RMIS.
Pharmacological regulatory network of key targets in TCM
We proposed the active components corresponding to the five key targets obtained above, constructed a drug-active component-key target gene network, and used Cytoscape for visualization (Fig. 7). The network contains eight herbs in YQAEF (Lonicerae Japonicae Flos, Forsythiae Fructus, Pogostemon Cablin Benth, Hedysarum Multijugum Maxim, Atractylodes Macrocephala Koidz, Saposhnikoviae Radix, Platycodon Grandiforus, and Licorice), 11 active pharmaceutical components, five key targets, and a total of 56 relationship pairs.
Verification with molecular docking
We selected the key core targets ACE, GLA, MMP2, SERPINE1, and MPO in the key signaling pathway obtained in the previous steps, and carried out molecular docking analysis with the main active components of YQAEF. We used the PubChem database to download the 3D conformers of quercetin, acetic acid, and raffinose, and the PDB database to download the IDs of MPO, MMP2, ACE, SERPINE1, and GLA, which are 6AZP, 1RTG, 2XY9, 1A7C, and 1NL0, respectively. The docking results showed better binding energy (Fig. 8A-E).
Quercetin formed a hydrogen bond with the MPO receptor target protein through the amino acid residues ASP-260, MET-253, and ARG-499 (Fig. 8A). The docking affinity between the active molecule and the protein was -9.7 kcal/mol. Quercetin formed a hydrogen bond with the MMP2 receptor target protein through the amino acid residues ASN-573, ALA-479, GLN-480, and GLU-525 (Fig. 8B). The docking affinity between the active molecule and the protein was -9.2 kcal/mol. Acetic acid formed a hydrogen bond with the ACE receptor target protein through the amino acid residues SER-147, HIS-353, and CYS-352 (Fig. 8C). The docking affinity between the active molecule and the protein was -3.5 kcal/mol. Acetic acid formed a hydrogen bond with the SERPINE1 receptor target protein through the amino acid residues TYR-37 and SER-41 (Fig. 8D). The docking affinity between the active molecule and the protein was -3.6 kcal/mol. Raffinose formed a hydrogen bond with the GLA receptor target protein through the amino acid residues LEU-136, SER-138, ASP-139, GLN-168, SER-169, and THR-183 (Fig. 8E). The docking affinity between the active molecule and the protein was -7.1 kcal/mol. These ligand compounds were well embedded in the active pocket of the receptor target protein.
Experimental validation in vivo
WB analysis of key targets proteins in UAID mouse oral cavity and larynx tissuesIgA and pIgR in mucosal epithelia play an important role in preventing pathogen adhesion to host cells, thereby blocking dissemination and further infection[26]. Seven days after the intervention, the oral cavity and larynx tissues of mice in each group were collected for WB examination. Although IgA and pIgR were normally expressed in the oral cavity and larynx tissues of the control group, their expression in the model group was significantly lower than that in the control group (P<0.001). The expression of IgA and pIgR in the YQAEF group was significantly lower than that in the model group (P<0.001), but was still significantly lower than that in the control group (P<0.05 or P<0.01, Fig. 9A-C).
In addition, among the five key targets in the network pharmacology analysis, compared with the control group, the expression of ACE and MPO proteins in the model group was significantly increased (P<0.001), while the expression of ACE and MPO in the YQAEF group was significantly lower than that in the model group (P<0.001), but was still significantly higher than that in the control group (P<0.001); the expression of GLA protein in the model group and the YQAEF group was significantly decreased (P<0.001) and the two groups were comparable; the protein expression of MMP2 and SERPINE1 was comparable among the three groups, with no statistical difference (Fig. 9D-I).
IHC detection of ACE and ACE2 proteins in UAID mouse oral cavity and larynx tissues
Seven days after the intervention, the oral cavity and laryngeal tissues of mice in each group were collected for IHC examination. The expression of ACE and ACE2 was observed in the nucleus and cytoplasm of the epithelial cells of the oral cavity and larynx, respectively. There was a small amount of ACE and ACE2 expression in the oral cavity and laryngeal tissue of mice in the control group. The expression of ACE and ACE2 in the model group was significantly higher than that in the control group (P<0.05, P<0.01), while the ACE expression in the YQAEF group was lower than that in the model group, and the ACE2 expression in the YQAEF group was significantly lower than that in the model group (P<0.05, see Fig. 10A-B). The above results indicated that the expression of ACE and ACE2 was elevated in upper airway immune dysfunction. In mice, YQAEF regulates the overexpression of ACE/ACE2 and improves upper airway immune dysfunction.
Discussion
Maintaining the immune function of the respiratory mucosa can effectively prevent respiratory diseases, and TCM is an effective alternative therapy[27]. In this study, we applied network pharmacology methods to discover the key active components of YQAEF in the regulation of RMIS. Our results showed that quercetin, acetic acid, and raffinose are important components in the regulation of RMIS, which may act on multiple pharmacological targets for virus infection prevention and treatment.
In vitro experiments and some animal models have shown that quercetin, a polyphenol derived from plants, has a wide range of biological actions, including antiviral[28-29], anti-inflammatory, and anti-carcinogenic activities[30-31], as well as attenuating lipid peroxidation, platelet aggregation, and capillary permeability[31]. Acetic acid is a short-chain fatty acid that regulates the participation of innate immune cells in the immune system, such as macrophages, neutrophils, and dendritic cells. It can regulate the differentiation of T cells and B cells and their mediated antigen-specific adaptive immunity, as well as provide energy for the body and other active roles, such as anti-inflammatory and antiviral activities[33-34]. Raffinose promotes the proliferation of bifidobacteria, improves the stomach, protects the liver, lowers blood cholesterol, enhances immunity, and has anti-cancer and anti-oxidation properties[35-36].
The potential target genes and signaling pathways of YQAEF in the regulation of the RMIS were predicted based on network pharmacology. Our results showed that ACE, GLA, MMP2, SERPINE1, and MPO are potential key targets for RMIS treatment. In addition, key target genes are involved in signal transduction, viral infection, endocrine resistance, and other related pathways involved in the regulation of RMIS.
Molecular docking is mainly used to study interactions between molecules. The more stable the binding conformation of the ligand and the receptor, the stronger the binding and the lower the energy[37]. The screened active components and key targets were verified by molecular docking technology. According to the optimal composite structure of key targets and active components, quercetin has better binding ability with MPO and MMP2. In addition, acetic acid has good binding ability with ACE and SERPINE1, and mullein has good binding ability with raffinose. These data suggested that the key chemical components of YQAEF have good binding activity with key targets in diseases.
Some homology exists between ACE2 and ACE[38]. The first step in SARS-COV-2 entry is the binding of the viral trimeric spike protein to the human receptor ACE2. Yan et al. presented the structure of human ACE2 in complex with a membrane protein, such as the chaperone, B0AT1. In this complex, ACE2 is a dimer. Furthermore, the receptor-binding domain of SARS-CoV-2 can interact with ACE2, implying that two trimeric spike proteins may bind to an ACE2 dimer. These structures provide a basis for the development of therapeutics that target this crucial interaction[5].
Based on the preliminary research foundation of our group and the results of this study, the targets of ACE and ACE2 were selected for further experimental verification, which suggested that YQAEF inhibits the overexpression of ACE2 in AURI by regulating RMIS, which indirectly reflects its potential antiviral function.
This study used bioinformatics to analyze the mechanism of action of YQAEF in the regulation of the RMIS and selected key targets for experimental verification in vivo. The results showed that the identical compound of YQAEF may regulate different targets, and ACE and ACE2 targets may interfere with BP and signaling pathways associated with respiratory diseases, such as COVID-19. This reflects the combined effect of multi-pathway and multi-target YQAEF. This study provides a scientific basis for YQAEF in the regulation of the RMIS and a new direction for exploring the potential antiviral mechanism of YQAEF.
References
[1] POURHOSEINGHOLI MA, RAHIMI-BASHAR F, VAHEDIAN-AZIMI A. Does context have a dramatic effect on results of mental health outcomes of ICU and non-ICU healthcare workers during the Coronavirus disease 2019 (COVID-19) outbreak[J]. Intensive and Critical Care Nursing, 2022: 103208.
[2] ZHANG T, LI X, CHEN Y, et al. Evidence mapping of 23 systematic reviews of Traditional Chinese Medicine Combined with western medicine approaches for COVID-19[J]. Frontiers in Pharmacology, 2021(12): 807491.
[3] LOU Z, ZHAO H, LU G. Mechanism and intervention of mucosal immune regulation based on "lung and large intestine being interior-exteriorly related" theory of traditional Chinese medicine[J]. Journal of Zhejiang University (Medical sciences), 2020, 49(6): 665-678. (in Chinese).
[4] HUANG W, JIANG B, LUO J, et al. Treatment of COVID-19 in Hemodialysis Patients Using Traditional Chinese Medicine: A Single-Center, Retrospective Study[J]. Frontiers in Pharmacology, 2022(13): 764305.
[5] YAN R, ZHANG Y, LI Y, et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2[J]. Science, 2020, 367(6485): 1444-1448.
[6] CUI XR, GUO YH, LIU QQ. Cangma Huadu granules, a new drug with great potential to treat coronavirus and influenza infections, exert its efficacy through anti-inflammatory and immune regulation[J]. Journal of Ethnopharmacology, 2022(287): 114965.
[7] TANG X, TONG L, GUO FF, et al. Analysis of potential role of Chinese classic prescriptions in treatment of COVID-19 based on TCMATCOV platform[J]. China Journal of Chinese Materia Medica. 2020, 45(13): 3028-3034. (in Chinese).
[8] LIU X, SHEN J, FAN D, et al. Yupingfeng San inhibits NLRP3 inflammasome to attenuate the inflammatory response in asthma mice [J]. Frontiers in Pharmacology, 2017(8): 944.
[9] XIAO M, TIAN J, ZHOU Y, et al. Efficacy of Huoxiang Zhengqi dropping pills and Lianhua Qingwen granules in treatment of COVID-19: A randomized controlled trial[J]. Pharmacological Research, 2020(161):105126.
[10] GUAN W, LAN W, ZHANG J, et al. COVID-19: antiviral agents, antibody development and traditional Chinese medicine[J]. Virologica Sinica, 2020, 35(6): 685-698.
[11] XIAO M, TIAN J, ZHOU Y, et al. Efficacy of Huoxiang Zhengqi dropping pills and Lianhua Qingwen granules in treatment of COVID-19: A randomized controlled trial[J]. Pharmacological Research, 2020(161): 105126.
[12] HU Y, LIU S, LIU W, et al. Potential molecular mechanism of Yishen Capsule in the treatment of diabetic nephropathy based on network pharmacology and molecular docking [J]. Diabetes, Metabolic Syndrome and Obesity, 2022(15): 943-962.
[13] HE D, HUANG JH, ZHANG ZY, et al. A network pharmacology-based strategy for predicting active components and potential targets of Liu Wei Di Huang Pill in treating type 2 diabetes mellitus[J]. Drug Design Development and Therapy, 2019(13): 3989-4005.
[14] RU J, LI P, WANG J, et al. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines[J]. Journal of Cheminformatics, 2014(6): 13.
[15] ZHANG GB, LI QY, CHEN QL, et al. Network pharmacology: a new approach for Chinese herbal medicine research[J]. Evidence-Based Complementary and Alternative Medicine, 2013(2013): 621423.
[16] BARSHIR R, FISHILEVICH S, INY-STEIN T, et al. GeneCaRNA: A comprehensive gene-centric database of human non-coding RNAs in the GeneCards suite[J]. Journal of Molecular Biology, 2021, 433(11): 166913.
[17] WANG Y, ZHANG S, LI F, et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics[J]. Nucleic Acids Research, 2020, 48(D1): D1031-1031D1041.
[18] YU G, WANG LG, HAN Y, et al. ClusterProfiler: An R package for comparing biological themes among gene clusters[J]. OMICS. 2012, 16(5): 284-287.
[19] Gene Ontology Consortium. Gene Ontology Consortium: Going forward[J]. Nucleic Acids Research, 2015, 43(Database issue): D1049-1056.
[20] CHEN L, ZHANG YH, WANG S, et al. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways[J]. PLoS One. 2017, 12(9): e0184129.
[21] OTASEK D, MORRIS JH, BOUAS J, et al. Cytoscape Automation: Empowering workflow-based network analysis[J]. Genome Biology, 2019, 20(1): 185.
[22] SZKLARCZYK D, GABLE AL, LYON D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets[J]. Nucleic Acids Research, 2019, 47(D1): D607-607D613.
[23] YANG JF, SHI SN, XU WH, et al. Screening, identification and validation of CCND1 and PECAM1/CD31 for predicting prognosis in renal cell carcinoma patients[J]. Aging (Albany NY). 2019, 11(24): 12057-12079.
[24] XIAN LI, LEI N , DUAN XH, et al., A mouse model of upper respiratory tract mucosal immunity dysfunction induced by cold stimulation[J]. Chinese Journal of Pathophysiology, 2011, 27(8): 1662-1664. (in Chinese).
[25] NOORI S, AL-AARAJI N, AL-SHAMERY E. Construction of dynamic protein interaction network based on gene expression data and quartile one principle[J]. Proteins. 2022, 90(5): 1219-1228.
[26] CORTHSY B. Role of secretory immunoglobulin A and secretory component in the protection of mucosal surfaces[J]. Future Microbiol. 2010, 5(5): 817-829.
[27] KURONO Y. The mucosal immune system of the upper respiratory tract and recent progress in mucosal vaccines[J]. Auris Nasus Larynx. 2022, 49(1): 1-10.
[28] LIU M, YU Q, XIAO H, et al. The Inhibitory activities and antiviral mechanism of medicinal plant component quercetin against grouper iridovirus infection [J]. Frontiers in Microbiology, 2020(11): 586331.
[29] LI Y, YAO J, HAN C, et al. Quercetin, inflammation and immunity[J]. Nutrients. 2016, 8(3): 167.
[30] XU D, HU MJ, WANG YQ, et al. Antioxidant activities of quercetin and its complexes for medicinal application [J]. Molecules. 2019, 24(6).
[31] BATIHA GE, BESHBISHY AM, IKRAM M, et al. The pharmacological activity, biochemical properties, and pharmacokinetics of the major natural polyphenolic flavonoid: Quercetin[J]. Foods. 2020, 9(3).
[32] LIU W, ZHANG M, FENG J, et al. The influence of quercetin on maternal immunity, oxidative stress, and inflammation in mice with exposure of fine particulate matter during gestation [J]. International Journal of Environmental Research and Public Health, 2017, 14(6).
[33] YAO Y, CAI X, FEI W, et al. The role of short-chain fatty acids in immunity, inflammation and metabolism[J]. Critical Reviews in Food Science and Nutrition, 2022, 62(1): 1-12.
[34] TROMPETTE A, GOLLWITZER ES, YADAVA K, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis[J]. Nature Medicine, 2014, 20(2): 159-166.
[35] LI X, HAI YW, MA D, et al. Fatty acid ester surfactants derived from raffinose: Synthesis, characterization and structure-property profiles[J]. Journal of Colloid and Interface Science, 2019(556): 616-627.
[36] HOSEINIFAR SH, HOSSEINI M, PAKNEJAD H, et al. Enhanced mucosal immune responses, immune related genes and growth performance in common carp (Cyprinus carpio) juveniles fed dietary Pediococcus acidilactici MA18/5M and raffinose[J]. Developmental and Comparative Immunology, 2019(94): 59-65.
[37] SONG X, ZHANG Y, DAI E, et al. Prediction of triptolide targets in rheumatoid arthritis using network pharmacology and molecular docking[J]. International Immunopharmacology, 2020(80): 106179.
[38] TIKELLIS C, THOMAS MC. Angiotensin-Converting Enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease [J]. International Journal of Peptide Research and Therapeutics, 2012(2012): 256294.
Editor: Yingzhi GUANGProofreader: Xinxiu ZHU

- 农业生物技术(英文版)的其它文章
- Optimization of Extraction Conditions for Total Flavonoids from Fructus Aurantii Immaturus and Its Anti-UVB Radiation Activity
- Research on Maize Seed Classification Method Based on Convolutional Neural Network
- Common Species Distribution Models in Biodiversity Analysis and Their Challenges and Prospects in Application
- Vegetable Tunnel House Technology in Tropical Island Countries
- Purification and Characterization of Hyaluronate Lyases Produced by Two Types of Bacteria
- Renewable Energy Seawater Desalination Technology and Application Analysis