
母细胞性浆细胞样树突细胞肿瘤2例临床病理研究
2023-09-04 03:27曹春育陈丽华戴桂红
安徽医药 2023年9期
曹春育,陈丽华,戴桂红
母细胞性浆细胞样树突细胞肿瘤(blastic plasmacytoid dendritic cell neoplasm,BPDCN)是一种罕见的高度侵袭性淋巴造血系统肿瘤,它起源于浆细胞样树突细胞的前体[1],特征性地表达白细胞分化抗原4(CD4)、白细胞分化抗原56(CD56)和白细胞分化抗原123(CD123),不表达T/B细胞、自然杀伤细胞和髓系特异性标记。该肿瘤在临床特征、组织学形态及免疫表型上与其他多种淋巴造血系统恶性肿瘤有重叠,故诊断较困难。本研究通过分析和观察2例BPDCN的临床表现、组织学形态及免疫表型,并结合相关文献复习,探讨其诊断与鉴别诊断要点。
1 资料与方法
1.1 临床资料 2例BPDCN分别为泰州市人民医院2020年住院病例及2019年会诊病例,本研究符合《世界医学协会赫尔辛基宣言》相关伦理要求,病人签署知情同意书。病例1,男性,76岁,病人因发现左侧颧面部肿物1年余并逐渐增大,近期感生长速度加快就诊,查体左侧颧面部可见一肿块,4.0 cm×4.0 cm,颜色暗红,边界不清,稍突出皮肤表面,无明显破溃,质地中等偏硬,与周围组织粘连不明显,按压无疼痛,未及波动感,周围皮肤无明显红肿(图1A),临床诊断为面部血管瘤行手术切除。病例2,男性,42岁,病人因发现左颈部包块1月入院,查体左颈部触及一淋巴结,长径2.0 cm,活动度可,无压痛,表面皮肤无红肿破溃,入院后行淋巴结活检(图1B)。
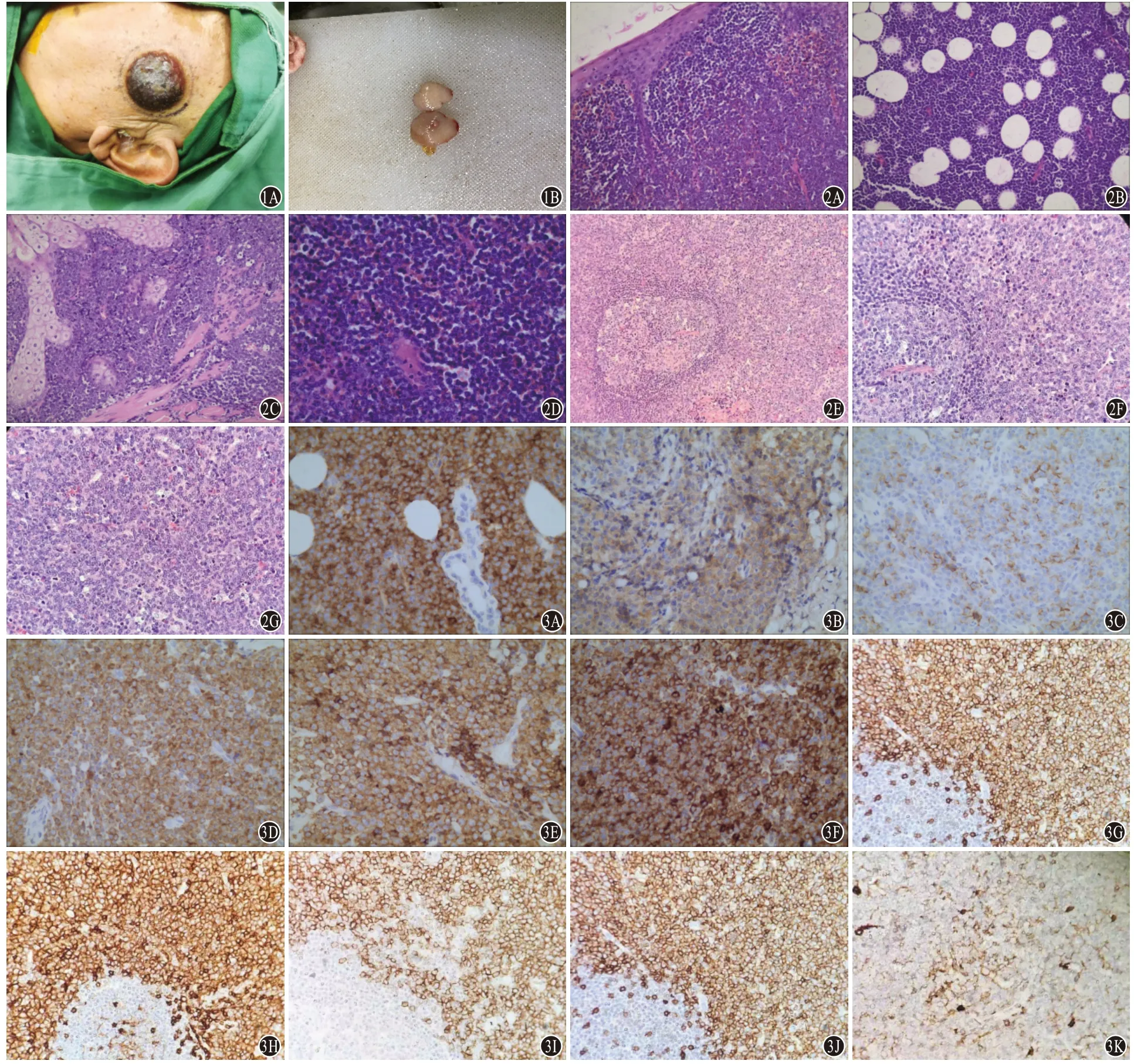
图1 母细胞性浆细胞样树突细胞肿瘤(BPDCN)2例临床皮损照片:1A为病例1左侧颧面部4.0 cm×4.0 cm暗红肿物,边界不清,稍突出皮肤表面,无明显破溃,质地中等偏硬;1B为病例2皮下直径2.0 cm结节,切面实性,灰红色,质地嫩 图2 母细胞性浆细胞样树突细胞肿瘤(BPDCN)2例皮损组织病理:2A为病例1肿瘤细胞弥漫分布于真皮内,肿瘤细胞与表皮之间无明显分界,但未侵犯表皮(HE染色×40);2B为病例1肿瘤细胞浸润皮下脂肪组织,可见“星空”现象(HE染色×40);2C为病例1肿瘤细胞围绕皮脂腺,在胶原纤维束之间浸润性生长(HE染色×200);2D为病例1肿瘤细胞中等大小,胞质稀少,核圆形、卵圆形或略不规则,染色质细腻,可见小核仁(HE染色×400);2E为病例2淋巴结结构消失,残留个别淋巴滤泡,肿瘤细胞弥漫性一致浸润,无明显间质反应(HE染色×100);2F为病例2肿瘤细胞沿副皮质区生长,包绕淋巴滤泡(HE染色×200);2G为病例2肿瘤细胞中等大小,胞质稀少,细胞核大,卵圆形或不规则,可见一小核仁以及核分裂象(HE染色×400) 图3 母细胞性浆细胞样树突细胞肿瘤(BPDCN)2例皮损组织免疫组织化学:3A为病例1肿瘤细胞白细胞分化抗原(CD)123阳性(EnVision法×400);3B为病例1肿瘤细胞CD4阳性(EnVision法×400);3C为病例1肿瘤细胞CD56阳性(EnVision法×400);3D为病例1肿瘤细胞B细胞淋巴瘤-2(Bcl-2)阳性(EnVision法×400);3E为病例1肿瘤细胞CD2阳性(EnVision法×400);3F为病例1肿瘤细胞CD7阳性(EnVision法×400) ;3G为病例2肿瘤细胞CD123阳性(EnVision法×400);3H为病例2肿瘤细胞CD4阳性(EnVision法×400);3I为病例2肿瘤细胞CD56阳性(EnVision法×100);3J为病例2肿瘤细胞CD43阳性(EnVision法×400);3K为病例2肿瘤细胞CD38阳性(EnVision法×400)
1.2 方法 送检组织经10%中性福尔马林固定,常规脱水,石蜡包埋,3 μm厚连续切片,分别进行苏木精-伊红(HE)染色、免疫组织化学和原位杂交。免疫组织化学染色采用EnVision二步法,所选一抗CD123、CD4、CD56、白细胞分化抗原43(CD43)、白细胞分化抗原33(CD33)、白细胞分化抗原20(CD20)、白细胞分化抗原79a(CD79a)、白细胞分化抗原10(CD10)、白细胞分化抗原2(CD2)、白细胞分化抗原3(CD3)、白细胞分化抗原5(CD5)、白细胞分化抗原7(CD7)、白细胞分化抗原8(CD8)、白细胞分化抗原30(CD30)、白细胞分化抗原38(CD38)、白细胞分化抗原21(CD21)、末端脱氧核苷酸转移酶(TdT)、白细胞分化抗原117(CD117)、髓过氧化物酶(MPO)、穿孔素、T淋巴细胞内抗原1(TIA-1)、颗粒酶B、白细胞分化抗原68(CD68)、白细胞分化抗原163(CD163)、白细胞分化抗原1a(CD1a)购自福州迈新生物技术有限公司,细胞角蛋白20(CK20)、突触素、嗜铬素A、B细胞淋巴瘤-2(Bcl-2)基因、B细胞淋巴瘤-6(Bcl-6)基因、转录因子C-myc、细胞周期蛋白D1(cyclin D1)、SRY相关高迁移率族盒蛋白-11(SOX-11)、S-100蛋白、肌酸激酶、上皮膜抗原(EMA)、间变性淋巴瘤激酶(ALK)、多发性骨髓瘤原癌基因-1(MUM-1)、白细胞共同抗原(LCA)、配对盒基因-5(PAX-5)、细胞核增殖相关抗原67(Ki-67)以及EnVision试剂盒均购自丹麦 DAKO 公司。EB病毒编码的小RNA(EBER)原位杂交试剂盒购自北京中杉金桥生物技术有限公司。免疫组织化学和原位杂交染色步骤按照试剂说明书进行,所有染色均设阳性对照和阴性对照。具体操作步骤按说明书进行。
2 结果
2.1 大体检查 病例1,送检带皮肿物1枚,大小4.0 cm×4.0 cm×1.2 cm,表面暗红色,无破溃,剖面灰红色、实性、质嫩。病例2,送检结节1枚,长径2.0 cm,剖面灰红色、实性、质嫩。
2.2 镜下观察 病例1,皮肤肿物示表皮萎缩变薄,真皮(图2A)及皮下胶原纤维间、脂肪组织内(图2B)见弥漫成片的肿瘤细胞浸润,并包绕皮脂腺、汗腺及神经生长(图2C),部分区域皮肤附属器破坏减少,无明显坏死和嗜血管现象,肿瘤细胞紧邻表皮,与表皮之间缺乏无细胞浸润带,真皮浅层见大量红细胞外渗。肿瘤细胞呈中等大小,形态较一致,胞质稀少,细胞核圆形、卵圆形或不规则,染色质细腻或空淡,可见明显的单个嗜酸性小核仁(图2D),核分裂易见,细胞凋亡多见,出现巨噬细胞吞噬碎片的“星空”现象。病例2,淋巴结活检示淋巴结结构破坏,局部残留少量的淋巴滤泡(图2E),瘤细胞呈弥漫性一致分布或沿副皮质区生长,包绕淋巴滤泡(图2F),肿瘤细胞间无背景细胞浸润和纤维间质反应,无血管增生。肿瘤细胞中等大小,介于中心母细胞和套细胞之间,胞质稀少,细胞核卵圆形或轻度扭曲呈角,染色质相对细腻,可见小核仁,核分裂易见(图2G)。
2.3 免疫组织化学染色及原位杂交结果 病例1:肿瘤细胞CD123(图3A)、CD4(图3B)、CD56(图3C)、Bcl-2(图3D)、CD2(图3E)、CD7(图3F)、CD43、CD33、CD10、C-myc均阳性,TdT、CD117、MPO、CD20、CD79a、Bcl-6、CD3、CD5、CD8、穿孔素、TIA-1、颗粒酶B、cyclin D1、SOX-11、S-100、CD38、CK20、突触素、嗜铬素A、CD163、CD68、CD1a、CD30、ALK、肌酸激酶、EMA均阴性;Ki-67阳性率约60%;原位杂交检测EBER阴性。病例2:肿瘤细胞CD123(图3G)、CD4(图3H)、CD56(图3I)、CD43(图3J)、CD38(图3K)、MUM-1均阳性;LCA、CD20、CD79a、PAX-5、CD10、C-myc、CD3、CD5、CD8、CD21、CD138、TdT、MPO、肌酸激酶均阴性;Ki-67阳性指数约40%;原位杂交检测EBER阴性。
2.4 病理诊断 2例均诊断为BPDCN。
2.5 随访 病例1病人术后2月,于原切口处皮下出现复发病灶,随后至上级医院就诊。病例2病人确诊后未进一步治疗,后失访。
3 讨论
BPDCN是一种罕见的血液系统恶性肿瘤,1994年由Adachi等[2]首次报道,由于其组织来源的不确定,根据其免疫表型曾被命名为无颗粒CD4+自然杀伤细胞白血病、母细胞性自然杀伤细胞白血病/淋巴瘤及无颗粒CD4+CD56+血液皮肤肿瘤。随后,陆续有研究证实该肿瘤起源于浆细胞样树突细胞(PDC)前体,在2008年世界卫生组织(WHO)造血与淋巴组织肿瘤分类中将其正式命名为BPDCN,归类为急性髓系白血病(AML)和相关肿瘤。2016年WHO修订分类中又将其作为一个单独类型归为髓系恶性肿瘤[1,3]。BPDCN的发病率极低,在血液恶性肿瘤中约占0.44%,在皮肤造血系统肿瘤中约占0.7%[4]。该肿瘤可发生于任何年龄,但主要发生于老年男性病人,可累及全身多个部位,最常见的发病部位为皮肤,皮损可单发或多发,大小不等,形态多样,可表现为瘀斑样、斑块或结节[5]。皮肤以外常见受累的部位包括淋巴结、脾脏,还有一些少见部位也有报道,如中枢神经系统、眼睛、鼻腔、扁桃体、肝肺、睾丸等,60%~90%的病例在诊断时发生骨髓和外周血受累的情况[6-8]。极少部分病人以急性白血病为首发特征,发病开始即累及全身[9]。本研究病人均为男性,病例1以皮肤受累为首发症状,表现为面部的暗红色结节,确诊后病人行骨髓活检及PET-CT全身检查,未发现骨髓和其他部位受累;病例2以淋巴结受累为首发症状,确诊时未发现皮肤病变及外周血受累。
BPDCN组织学特征是中等大小的肿瘤细胞呈弥漫性浸润,细胞核单个,圆形、卵圆形或不规则,染色质细腻,可见1个或多个嗜酸性小核仁,胞质稀少,类似于母细形态,核分裂象数量不等。发生于皮肤时,肿瘤细胞主要累及真皮层,也可侵及皮下脂肪组织,但不侵犯表皮,肿瘤细胞与表皮之间常见无细胞浸润带,少数病例也可缺乏,一般无肿瘤性坏死及血管壁侵犯。本研究病例1肿瘤细胞与表皮之间缺乏无细胞浸润带,有显著的红细胞外渗,核分裂象及凋亡多见,形成“星空”现象。累及淋巴结时,淋巴结结构破坏,可见残留淋巴滤泡,肿瘤细胞在滤泡间区或髓质区弥漫性浸润,本研究病例2肿瘤细胞呈弥漫性或副皮质区浸润,淋巴结内无背景细胞浸润和纤维间质反应、无血管增生。累犯骨髓时可表现为微小的间质浸润或局部形成大片结节,骨髓涂片中肿瘤细胞原始,缺乏颗粒,胞质不规则呈伪足样突起,形成“手镜样”形态,细胞膜和伪足可见胞质空泡。
BPDCN诊断主要依赖免疫组织化学,肿瘤细胞特征性表达CD4、CD56和PDC相关标记[CD123、白细胞分化抗原303(CD303)、CD2相关蛋白(CD2AP)、T细胞淋巴瘤1(TCL1)等],除了CD2、CD7(T细胞标记)和CD33(髓系标记)[6],一般不表达T细胞(CD3、CD5)、B细胞[白细胞分化抗原19(CD19)、CD20和CD79a]、自然杀伤细胞(颗粒酶B、穿孔素和TIA-1)和髓系(MPO、CD13和CD117)相关标记。部分病例可表达TdT、CD10、Bcl-2、MUM-1、CD68、S-100[1,10]。EBER原位杂交检测阴性。本研究2例免疫组织化学CD4、CD56、CD123和CD43均为阳性;例1表达CD33、CD10、Bcl-2及T细胞标记CD2、CD7;例2表达CD38和MUM-1;2例TdT及其他T/B细胞标记、细胞毒性标记及髓系标记均为阴性;2例原位杂交检测EBER阴性。BPDCN病人无特异性的遗传学改变,约一半以上病例会出现染色体异常,常发生改变的染色体包括5q、6q、13q、12p和9单体、15单体。基因表达谱分析发现BPDCN存在多种基因异常,包括抑癌基因的失活[视网膜母细胞瘤蛋白1(RB1)、肿瘤抑制基因P53(TP53)、细胞周期蛋白依赖性激酶抑制因子1B(CDKN1B)、细胞周期蛋白依赖性激酶抑制因子2A(CDKN2A)],癌基因的激活(KRAS基因、NRAS基因、HES6基因、RUNX2基因、FLT3基因),表观遗传调节因子的突变[DNA羟甲基化酶10-11转位基因2(TET2)、DNA羟甲基化酶10-11转位基因1(TET1)、DNA甲基化转移酶3A(DNMT3A)、异柠檬酸脱氢酶1(IDH1)、异柠檬酸脱氢酶2(IDH2)]以及NF-κB通路[Bcl-2和干扰素调节因子4(IRF4)]的异常激活等,但这些改变对诊断该疾病并没有特异性[11-12]。
BPDCN鉴别诊断包括:(1)髓系肉瘤(MS)或白血病[AML和慢性粒-单核细胞白血病(CMML)]。两者临床表现、细胞学形态和免疫表型上均有重叠,AML和CMML可表达TdT、CD4、CD56,AML中亦可表达CD123和TCL1,但髓系肉瘤(MS)或白血病(AML和CMML)同时表达髓系标记MPO、CD117、CD34可排除。(2)Burkitt淋巴瘤。好发于儿童和年轻人,常发生于结外部位,最常累及颌骨,而BPDCN主要发生于老年病人,易累及皮肤。形态学上两者均表现为单一的幼稚母细胞样,均可出现“星空”现象,但前者瘤细胞核染色质呈粗块状,后者染色质相对细腻。免疫组织化学前者表达CD20、CD79a、Bcl-6阳性,不表达TdT、CD123、CD4、CD56、Bcl-2,Ki-67指数可高达90%~100%,可与BPDCN鉴别。(3)淋巴母细胞淋巴瘤(LBL)。BPDCN可发生于儿童病人,肿瘤细胞形态呈母细胞样,可见“星空”现象,且可表达TdT和CD7,而少数LBL可表达CD4和CD56,故诊断时两者需鉴别。LBL主要累及儿童及青年,T-LBL主要表现为纵隔肿块,B-LBL主要累及淋巴结,免疫组织化学TdT呈弥漫一致的强阳性,同时表达T细胞(CD2、CD3、CD5)或B细胞(CD20、CD79a、PAX-5)相关抗原,不表达CD123可鉴别。(4)套细胞淋巴瘤母细胞样变异型。与BPDNC形态学有交叉,瘤细胞均表现为母细胞样,形态一致,核圆形或有扭曲,染色质细腻,核分裂象多见,可见“星空”现象,但前者免疫组织化学CD5、cyclin D1、SOX11阳性可鉴别之。(5)结外自然杀伤细胞/T细胞淋巴瘤(鼻型自然杀伤细胞/T细胞淋巴瘤)。可表达CD4或CD56,或两者同时表达,但其肿瘤细胞呈多形性,不具有母细胞形态,且具有明显的坏死和嗜血管特征,免疫表型T细胞标志[CD3、T细胞活化连接蛋白(LAT)、Zeta链相关蛋白激酶70(ZAP-70)]和细胞毒分子(穿孔素、TIA-1)阳性表达,原位杂交EBER阳性,有助于鉴别。(6)蕈样霉菌病。组织学特征为真皮内小到中等大小的脑回状肿瘤细胞呈灶性、片状或弥漫性浸润,可累及皮下脂肪组织,可见亲表皮现象及Pautrier微脓肿,免疫组织化学肿瘤细胞虽然可以表达CD4,但不表达CD56、CD123。(7)Langerhans细胞组织细胞增生症。免疫组织化学表达CD4、CD56及CD123,与BPDCN有重叠,但前者同时表达CD1a、Langerin、S-100,瘤细胞核更加不规则,具有丰富的嗜酸性胞质,肿瘤背景中通常出现嗜酸性粒细胞。(8)Merkel细胞癌。发生于皮肤,瘤细胞具有母细胞样外观,且免疫组织化学表达CD56,可能与BPDCN相似,但前者还表达神经内分泌标记(嗜铬素A、突触素)和CK20(核周点状阳性)。(9)小圆细胞性恶性黑色素瘤。肿瘤细胞形态一致,小圆形、胞质稀少、胞核深染,圆形或卵圆形,可见核仁,形态类似淋巴母细胞,免疫组织化学MelanA、HMB45、S-100、SOX-10均阳性表达,可与BPDNC相鉴别。
BPDCN侵袭性强,进展较快,预后较差,中位生存时间约1年[5-6],目前对该病尚无统一的最佳治疗方案,多数采用AML型、急性淋巴细胞白血病(ALL)型或淋巴瘤型化疗方案[6]并结合自体或异体干细胞移植(SCT)等巩固治疗。单纯放疗对BPDCN病人无效,但它可以增强化疗的效果,特别是对皮肤有病变的病人[13]。BPDCN靶向治疗的研究正在进行中,针对CD123的靶向药物SL-401是第一个专门批准用于治疗成人及2岁儿童以上BPDCN的药物[14],此外,CD123嵌合抗原受体(CAR)T细胞(CAR-T)[15]以及Bcl-2抑制剂venetoclax也已被用于治疗化疗耐药的BPDCN病人[16],但具体疗效仍需大量及长期的临床试验来验证。
总之,由于BPDCN罕见,且组织学形态不特异,免疫表型不典型,与多种淋巴造血系统肿瘤有重叠,使BPDCN的诊断特别具有挑战性,其正确诊断必须在排除其他相似肿瘤的基础上,结合临床表现、组织形态特征及免疫表型综合分析,必要时尚需加做基因检测协助诊断。
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
传染病信息(2022年3期)2022-07-15
国际放射医学核医学杂志(2021年10期)2021-02-28
中国临床医学影像杂志(2019年1期)2019-04-25
中国生殖健康(2019年9期)2019-01-07
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中华老年多器官疾病杂志(2016年9期)2016-04-28
中国组织化学与细胞化学杂志(2016年3期)2016-02-27
磁共振成像(2015年5期)2015-12-23
天津医科大学学报(2015年3期)2015-06-05
