
共存网络分析揭示抑郁症患者肠道微生物改变
2023-09-01 01:59:50陈棉棉王培琳谢新晖聂昭雯刘忠纯
神经损伤与功能重建 2023年8期
陈棉棉,王培琳,谢新晖,聂昭雯,刘忠纯
0 背景
抑郁症(major depressive disorder,MDD)发病率持续增高,影响患者的身心健康[1]。MDD 相关的遗传、神经内分泌和免疫相关机制受到广泛关注[2]。随着测序技术的发展,肠道微生物群与MDD 的关系逐渐成为研究热点[3,4]。在抑郁症患者中检测到了肠道微生物群的改变[5],将MDD患者的肠道微生物移植到啮齿类动物体内,可以诱发抑郁样行为[6,7],口服益生菌后又观察到此类抑郁样行动的减少[8]。因此,肠道微生物群被认为与MDD的致病机制相关。
然而,目前对肠道微生物群落的分析主要集中在疾病中微生物物种丰度或功能的改变,较少关注对物种间关系的探索,可能导致对微生物群落部分特征的忽略。在群落中,其组成物种不是独立的,而是相互作用的。在宏观生态研究,例如对海洋、土壤等环境微生物群落的研究中,已经广泛开展了微生物共存网络(co-occurrence network)的分析,以获取微生物相互作用、结构变化的信息[9,10]。而在肠道微生物领域,共存网络分析仍然有待发展。
因此,本研究招募MDD 患者,采集粪便样本进行宏基因组测序,将数据应用于微生物共存网络的构建与后续的分析。构建肠道微生物共存网络,探索与健康对照相比,MDD 人群肠道微生物共存网络的改变,并比较2 组网络的拓扑属性以及关键节点的变化,以补充一个新的、纯丰度或功能数据以外的视角,探索MDD 中微生物物种之间相互作用关系的改变,探明MDD患者失调的肠道微生态。
1 资料与方法
1.1 一般资料
从2020 年至2021 年就诊于武汉大学人民医院精神卫生中心的患者中,招募首发或复发MDD 患者,符合《精神障碍诊断与统计手册(第五版)》(Diagnostic and Statistical Manual of Mental Disorders-fifth edition,DSM-5)中重性抑郁发作(当前2 周)的诊断标准。同时招募一般人口学数据相匹配的健康人纳入对照组(healthy control,HC)。排除患有或共病符合DSM-5标准的其他精神障碍以及脑器质疾病所致精神障碍,患有严重的躯体疾病、代谢相关疾病或者免疫相关疾病,既往颅脑外伤史,1周内有益生菌、益生元、抗生素使用史的受试者。本研究通过武汉大学人民医院学术伦理道德委员会审核。所有被试均自愿参与本项研究并签署知情同意书。
1.2 方法
1.2.1 患者信息收集 为全面反映样本特征,采集一般人口学信息,并使用汉密尔顿抑郁量表(Hamilton depression scale-17,HAMD-17)和患者健康问卷(Patient Health Questionnaire-9,PHQ-9)进行抑郁严重程度的他评和自评;使用汉密尔顿焦虑量表(Hamilton anxiety scale,HAMA)和广泛性焦虑障碍量表(Generalized Anxiexy Disorde-7,GAD-7)进行焦虑严重程度的他评和自评;患者健康问卷躯体症状群量表(Patient Health Questionnaire-15,PHQ-15)进行躯体症状严重程度的自评。粪便样本使用粪便DNA 样本保存管(江苏康为世纪)采集,利用布里斯托大便评分(Bristol Stool Form Scale,BSS)对患者提供的粪便样本进行质量控制[11]。
1.2.2 宏基因组分析 使用NEBNext®Ultra™DNA Library Prep Kit for Illumina 试剂盒(NEB,USA)构建测序文库,Illumina Hiseq2500 测序仪(Illumina,San Diego,CA,USA)用于测序,每个样本输出6 GB原始数据。Readfq 软件(version 8,https://github.com/cjfields/readfq)用于预处理以及去宿主,使用MEGAHIT 软件(version 1.0.4-beta)组装分析,通过Bowtie2(version 2.2.4,http://bowtiebio.sourceforge.net/bowtie2/index.shtml)软件将各样品Clean Data分别对比至其Scaftigs上。使用MetaGeneMark 软件(version 2.10,http://topaz.gatech.edu/GeneMark)进行ORF预测,CD-HIT[12]软件(version 4.5.8,http://www.bioinformatics.org/cd-hit/)去冗余。通过Bowtie2 软件将各样品Clean Data 比对至初始gene catalogue,计算得到各基因在各样品中的丰度信息。使用DIAMOND[13]软件(version 0.9.9.110, https://github.com/bbuchfink/diamond/),将Unigenes 与NCBI的NR数据库进行比对获得各个样品在各个分类层级上的丰度信息。
1.2.3 微生物共存网络分析 使用R语言ggclusternet包[14],基于Spearman 等级相关性矩阵构建种水平肠道微生物共存网络。由于低丰度物种数据可能导致计算中的假阳性,选取丰度前5%的物种数据用于后续计算分析之中。并对微生物共存网络进行可视化,其中物种用节点表示,物种之间的相关性用连接节点的边表示。R语言ggclusternet包同时用于计算网络拓扑学参数,包括连接性、平均度、平均路径长度、直径、聚集系数等,并计算节点包括Zi(模块内连通度的Z-score)和Pi(模块间连通度的参与系数)值等相关属性。使用R 语言psych 包(version 2.2.5)对关键节点的相对丰度与一般人口学数据及临床量表评估结果进行Spearman相关分析,使用pheatmap包(version 1.0.12)进行可视化呈现。
1.3 统计学处理
使用SPSS 25.0 进行统计分析。本研究中年龄、BMI值等连续变量用(±s)表示,组间比较使用独立样本均数t检验;分类数据以频数和百分比(%)表示,组间比较使用χ2检验;P<0.05为差异有统计学意义。
2 结果
2.1 一般人口学
本研究共纳入MDD组35例,HC组35例。2组的年龄、性别、体质量指数(body mass index,BMI)、粪便性状等差异无统计学意义;在抑郁和焦虑症状的严重程度上,MDD 组的自评及他评评分均显著高于HC 组(P<0.001),且具有更多的躯体不适症状(P<0.001),见表1、表2。
表1 2组一般资料比较[(±s)或例(%)]
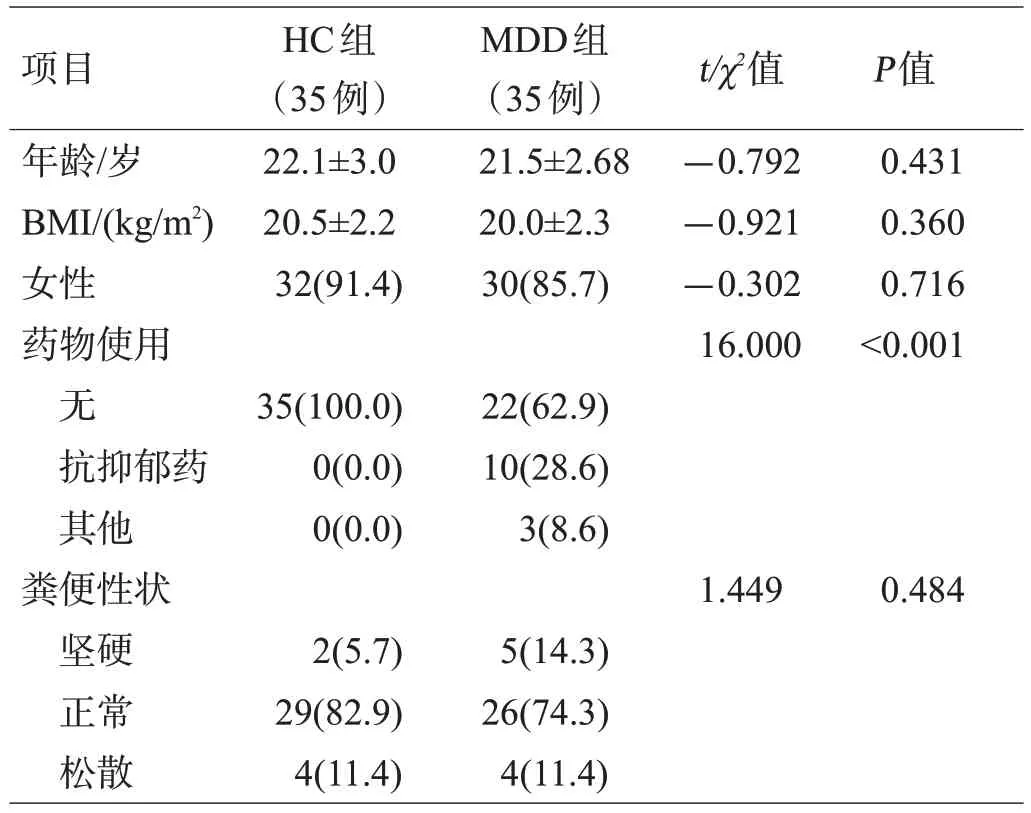
表1 2组一般资料比较[(±s)或例(%)]
项目年龄/岁BMI/(kg/m2)女性药物使用无抗抑郁药其他粪便性状坚硬正常松散HC组(35例)22.1±3.0 20.5±2.2 32(91.4)MDD组(35例)21.5±2.68 20.0±2.3 30(85.7)t/χ2值-0.792-0.921-0.302 16.000 P值0.431 0.360 0.716<0.001 35(100.0)0(0.0)0(0.0)22(62.9)10(28.6)3(8.6)1.449 0.484 2(5.7)29(82.9)4(11.4)5(14.3)26(74.3)4(11.4)
表2 2组各量表评分比较(分,±s)
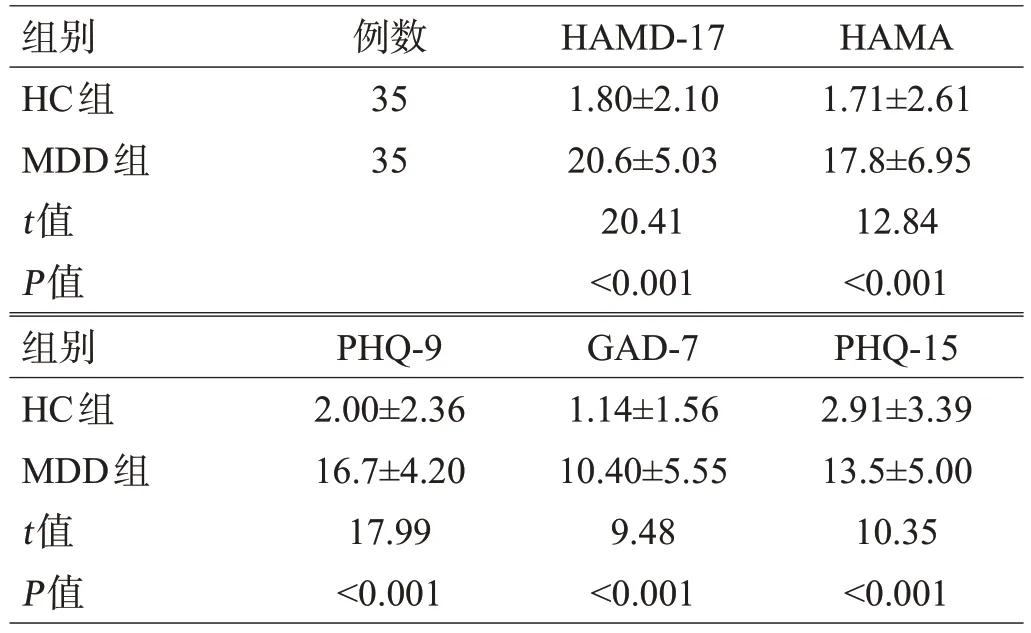
表2 2组各量表评分比较(分,±s)
组别HC组MDD组t值P值例数35 35 HAMD-17 1.80±2.10 20.6±5.03 20.41<0.001 HAMA 1.71±2.61 17.8±6.95 12.84<0.001组别HC组MDD组t值P值PHQ-9 2.00±2.36 16.7±4.20 17.99<0.001 GAD-7 1.14±1.56 10.40±5.55 9.48<0.001 PHQ-15 2.91±3.39 13.5±5.00 10.35<0.001
2.2 共存网络
为了描述肠道微生物群落中物种之间潜在关系,基于Spearman相关系数构建了2 组微生物共存网络,见图1。分布在2个共存网络中的物种主要来自4个门(Actinobacteria、Bacteroidetes、Firmicutes和Proteobacteria)。与负相关相比,2个网络的强正相关的数量要高得多,但HC组的微生物群落网络更为复杂。为了量化差异,计算了2 个微生物网络的拓扑特征,见表3。结果显示,与使用相同方法构造的HC网络相比,MDD网络显示出较少的节点数和连接数。此外,MDD中的平均度和图密度均较低,反映了MDD中物种之间的相互作用较少,网络较稀疏。2组共存网络拓扑结构复杂性不同,直径和平均聚集系数等参数均具有一定的差异。
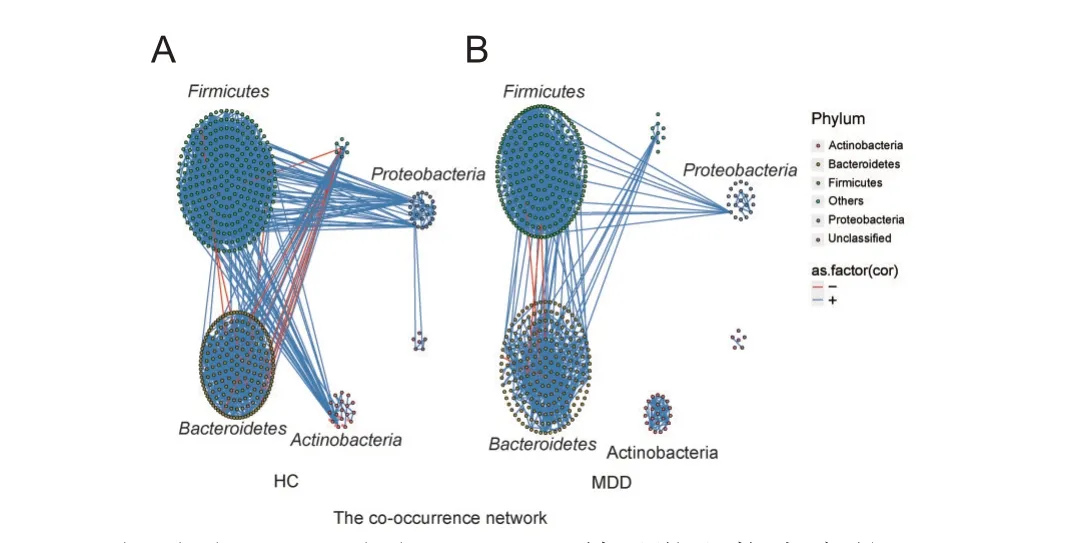
图1 2组受试者肠道微生物群落的共存网络图
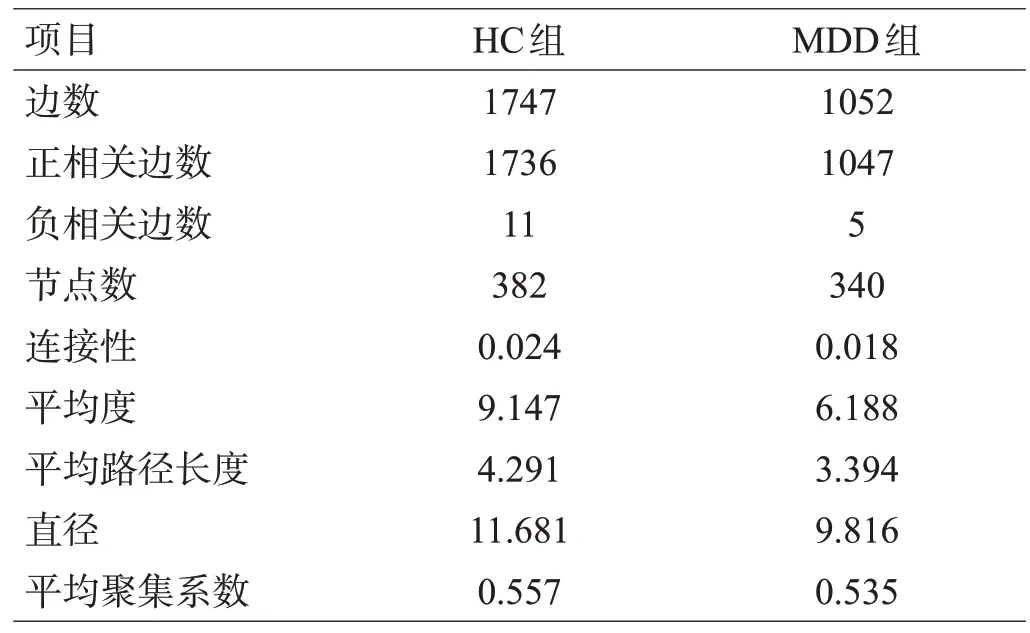
表3 2组的网络拓扑结构属性
2.3 随机网络
建立相同节点数以及相同连接数的Erdӧs-Réyni随机网络,发现HC组和MDD组网络的平均聚集系数极高,但平均路径长度较小,接近随机网络,表明2 组肠道网络具有小世界特性,其聚集相较随机网络更为显著,见表4。从单个节点度分布图上可以看出,2 组微生物的度分布遵循幂律分布规律,大部分的节点具有较低的度,只有少数核心节点具有较高的度,表明2组网络都是无标度网络,见图2。
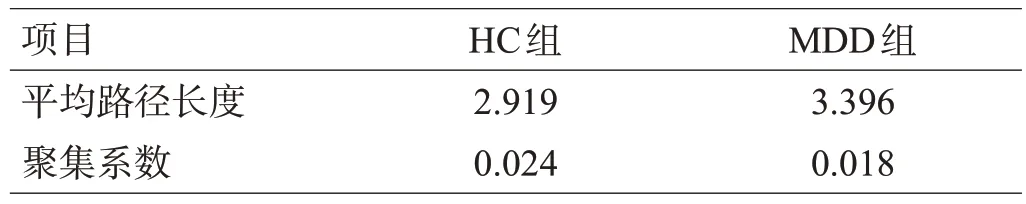
表4 2组的随机网络拓扑结构属性
2.4 Zi-Pi
为了研究MDD 网络中改变的拓扑结构是否由特定的生物驱动,本研究进一步测量了网络中每个物种的相对影响。依据节点的拓扑特征寻找网络核心节点是网络分析的热点。目前,基于Zi-Pi网络核心节点判别方法得到了广泛的应用[15]。模块是网络中高度连接的区域,是系统发育、进化或功能上独立的单元,模块中心节点(Module hubs,模块内部具有高连通度节点,Zi >2.5且Pi<0.62)作为特定模块内复杂交互网络的关键物种,对维持这一协同单元的正常功能有重要作用,连接节点(Connectors,模块之间高连通度节点,Zi<2.5且Pi>0.62)被认为可以维持网络的一致性[16]。我们发现在HC网络中,Ruminococcus flavefaciens是模块中心节点,Clostridium CAG 389和Lachnospiraceae bacterium.A2是连接节点;MDD 网络中uncultured Blautia sp,Bacteroides mediterraneensis和Parabacteroides distasonis是MDD网络的模块中心节点,见图3。
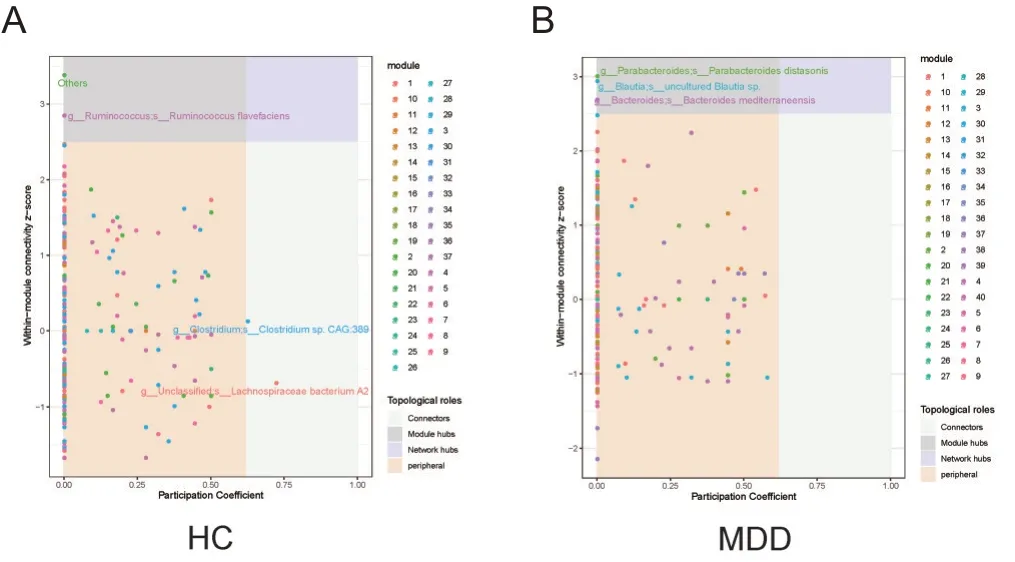
图3 2组网络节点的Zi-Pi图
2.5 相关热图
根据Zi 和Pi 的网络核心节点判别方法寻找到关键物种后,对其丰度与临床量表评估得分进行了Spearman相关分析,发现在经过FDR 校正后,Ruminococcus flavefaciens丰度与临床量表HAMD-17(ρ=-0.265,P=0.026)、PHQ-9(ρ=-0.260,P=0.030)、GAD-7(ρ=-0.315,P=0.008)得分之间存在显著相关;Clostridium CAG 389丰度与临床量表HAMD-17(ρ=-0.326,P=0.006)、HAMA(ρ=-0.309,P=0.009)、PHQ-9(ρ=-0.327,P=0.006)、GAD-7(ρ=-0.312,P=0.009)以及PHQ-15(ρ=-0.340,P=0.004)得分之间存在显著相关,见图4。
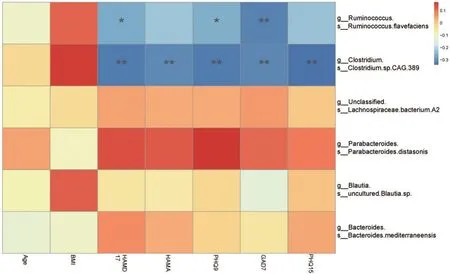
图4 关键物种丰度和临床指标的相关热图
3 讨论
目前针对MDD 患者肠道微生物物种丰度或功能改变进行的研究已经取得了一定成果,但对微生物相互作用关系的探索仍显欠缺。本研究使用共存网络分析,从另一个角度展示肠道微生态的变化,发现与HC组相比,MDD 组中微生物相互作用有所减少,群落中的关键成员改变,反映了MDD 组肠道群落微生态失调。
利用对微生物相关关系的认识,建立了共存网络,2组肠道微生物群落均表现出物种间较强正相关的共存网络。健康的肠道微生物组是一个复杂网络,适应不断变化的环境条件。在人类肠道微生物组的背景下,这些相互作用可能源自互补的资源获取策略、生态位划分或包括代谢物在内的资源转移[17]。与HC相比,MDD 组的网络拓扑属性发生了变化,网络较稀疏,物种之间的相互作用较少。而在MDD 中这些相互作用的减少可能会增加受到环境扰动时肠道微生态系统的不稳定。
核心微生物群在微生物群落中起着重要作用,并维持其功能。因此,核心微生物的鉴定非常重要。但目前核心微生物的鉴定标准尚未统一[18]。基于Zi和Pi的网络核心节点判别方法在生态网络模块中所识别的关键节点,被认为对维持微生物群落结构稳定性可能起重要作用[16]。我们测量了网络中每个物种的相对影响。为了确定连接不同微生物、与其他微生物密切相关,能够影响网络中的其他节点的潜在关键物种,我们重点研究了基于Zi-Pi 网络核心节点判别方法寻找到的枢纽物种,并将其丰度与量表得分进行相关性分析,发现其中2个关键物种的丰度与临床症状严重程度呈负相关。
然而,由于目前对其他关键物种研究尚不足,我们着重关注已被相关研究报道的物种。Ruminococcus flavefaciens是HC 网络模块中心节点,Parabacteroides distasonis是MDD 网络的模块中心节点。瘤胃球菌属(Ruminococcus)在精神疾病中受到广泛关注。动物研究发现瘤胃球菌属的丰度与小鼠的快感缺乏行为之间存在正相关[19]。此外,补充益生元在抗抑郁同时也表现出了瘤胃球菌属丰度的降低[20]。另一项动物研究直接引入单一的瘤胃球菌物种,发现补充Ruminococcus flavefaciens能够消除度洛西汀的抗抑郁作用[21]。在我们的研究中,Ruminococcus flavefaciens的丰度与抑郁和焦虑症状呈负相关,而且在HC网络中被确认为模块中心节点,具有模块内的高连通度,表明该物种潜在的重要性。Ruminococcus flavefaciens在正常肠道微生态网络以及与情绪、行为的关系有待进一步探索。
Parabacteroides distasonis作为潜在的益生菌,对结直肠癌[22]和炎症性肠病[23]等具有保护作用,可以促进人类的消化健康。然而,相矛盾的是,在许多疾病模型中都发现了Parabacteroides distasonis的致病作用[24]。同样的,动物研究发现,Parabacteroides distasonis灌胃处理诱导小鼠产生抑郁样行为[25]。作为被发现的第1个具有诱导抑郁作用的特定菌种,结合我们的研究结果,Parabacteroides distasonis对于人体的潜在致病作用不容忽视。综上所述,网络分析有助于提供补充纯丰度或功能数据的视角,表明MDD中微生物相互作用关系和关键物种的改变,并进一步表明与对照相比肠道微生态失调。
本研究局限在于,数据为横断面数据,无法推测定向网络。需要纵向数据或合成群落的自下而上方法进一步探索菌株-菌株相互作用的方向性以及探索特定菌株对群落的影响。此外,我们的样本量较小,尽管纳入的主要为首发未用药患者,但对其具体病程及用药情况未做进一步的了解,未来有必要对更大的数据集进行进一步研究,进一步探索MDD患者肠道微生物物种相互作用关系的改变,与临床特征的联系,尤其是抑郁发作期和缓解期的纵向改变,为更进一步的实验提出合适的假设。
4 结论
本研究使用共存网络分析,探讨了MDD和健康人群肠道微生物的差异,发现肠道微生物组的复杂生态网络在MDD 中发生变化,微生物相互作用减少,群落中关键成员有所改变。对这些有所改变的相互关系以及关键物种对MDD 的影响值得进一步研究。未来在MDD 中进行的肠道微生物研究有必要在共存网络方面做更多的努力,从纯丰度或功能的单一角度之外探讨MDD中肠道微生物群落的改变。
