
一种嵌合式DNA 聚合酶的表达及功能验证
2023-08-08 04:03:18王玉玲张闻慧吴银荣赵梦然罗忠宜
安阳工学院学报 2023年4期
王 尧,王玉玲,张闻慧,吴银荣,赵梦然,罗忠宜,林 森
(1.安阳工学院 生物与食品工程学院,河南 安阳 455000;2.澳大利亚外科诊断有限公司,澳大利亚 悉尼 2069;3.安阳康圣环球医学检验所有限公司,河南 安阳 455000)
DNA 的组成成分是脱氧核糖核酸[1],DNA 聚合酶是DNA 合成的主要功能大分子,通过4 种不同的脱氧核苷酸导入单链模板DNA 实现副本DNA 合成。在上述过程中,单个核苷酸与模板DNA 链上的碱基通过DNA 聚合酶实现配对。通过该过程高保真聚合酶被广泛应用到病毒分子研究、分子诊断以及克隆和合成生物学等领域。
目前,常见的聚合酶中,Taq DNA 聚合酶是用于PCR 扩增的最常见的酶[2]。耐热性极强,在95℃下半衰期为40 min,扩增能力强,但保真性较差。Vent DNA 聚合酶的保真度比观察到的Taq DNA 聚合酶高5~15 倍。Thermo Scientific Phusion高保真DNA 聚合酶为高性能PCR 设定了黄金标准[3]。Tth DNA 聚合酶是一种具有内在逆转录酶活性的热稳定DNA 聚合酶[4]。Pfu DNA 聚合酶是一种从Pyrococcus furiosus 中分离出来的约90 kDa 的高保真、热稳定的酶。Phusion 高保真DNA聚合酶的误差率仅是Taq 的1/50,是Pfu 的1/6。除上述聚合酶外,Q5 高保真DNA 聚合酶为保真度和稳定性设定了新标准,其保真度放大率最高(比Taq 高约280 倍),Q5 具有超低的误差率。Q5 由一种新型、可热的DNA 聚合酶组成,该聚合酶融合到增强过程性的Sso7d DNA 结合结构域,提高了性能的速度、保真度和可靠性[5]。Q5 热启动DNA 聚合酶利用独特的aptamer 来增加热启动便利性,而无需添加激活步骤。高保真聚合酶精度的提高使它们比标准Taq 聚合酶更适合敏感的下游应用[6]。使用PCR 扩增产生DNA 进行测序时应首选高保真聚合酶,主要因为Taq 聚合酶在PCR 的扩增子中会引起不匹配,影响测序数据的分析。因此,高保真聚合酶比Taq 聚合酶的应用领域更广[7]。
本研究将通过尝试不同培养基、纯度和浓度等物理条件,快速和经济地将本实验室已经建立的嵌合DNA 聚合酶模板进行重组、表达与纯化,从而得到一种能比市面上一些低保真度的聚合酶更好、更快地测出所需要数据、有良好保真度和稳健性的高精确度聚合酶,降低实验过程中聚合酶的出错率,节约反应时间。
1 材料与方法
1.1 实验材料
1.1.1 嵌合DNA 聚合酶的基因合成
目的基因序列由南京金斯瑞生物科技有限公司合成,亚克隆至pET11a 载体(NdeI 和EcoRI酶切位点之间),载体携带氨苄西林(AMP)抗性,用于阳性菌株的筛选。
1.1.2 仪器设备
全自动台式快速蒸汽灭菌器、ZEY-2102C 37 度恒温箱、实用垂直单人净化工作台、-80℃超低温保存箱、4℃冰箱、THZ-82B 气浴恒温震荡器、自动紫外可见分光光度计、INFORS HT恒温箱、SHAEER 摇床、H4-21KR 落地式高速冷冻离心机、电子天平、ATS 仪器高压破碎、磁力搅拌器、OHAUS-PH 仪、Nano-300 Microspetrophotometer、SCIOGE 快速离心机、PCR 仪、DYY-12 型电泳仪及其他仪器设备。
1.1.3 主要试剂
50 mol/L Tris-HCl,2 mol/L EDTA,1 mol/L DTT,10 % 甘 油,300 mol/L NaCl,ddH20,Lysis buffer(pH=8),Elution buffer(pH=8),Washing buffer 1 (pH=8),Washing buffer 2 (pH=8),Binding buffer(pH=8)。
1.1.4 主要培养基配制
1 L 液 体LB 配 方:10 g Tryptone,5 g Yeast Extract, 10 g NaCl, 0.1 g Glucose。
200 mL 固体LB 配方:2 g Tryptone,1 g Yeast Extract,2 g NaCl, 0.02 g Glucose,2.6 g Agar。
1.2 实验方法
1.2.1 蛋白表达
超净台内把菌涂在含AMP 的固体LB 培养皿,封口膜封口,放在37℃恒温箱培养12~16 h。用移液枪枪头取单菌落放在新的含AMP 固体LB培养皿上,用灭菌后的接种环进行涂开(涂3~4遍)越密越好,封口膜封口,放37℃恒温箱培养12~16 h。在液体LB中加入AMP抗生素(1∶1000)混合均匀,灭菌接种环接菌在液体LB 中混匀,放在37℃恒温振荡器3 h 进行摇菌。取样到比色皿中,用自动紫外分光光度计测OD 值,约为0.6时加终浓度1 mol/L IPTG 开始诱导。①28℃、120 r/min、37℃摇床6 h ②20℃ 200 r/min 37℃24 h。诱导后的样品用750 mL 大离心管离心4℃、4 000 r/min、15 min。离心前取200 μL,离心后取5 μL 加20 μL 上样缓冲液,剩下的分装到50 mL 的离心管再次离心(4℃、4 000 r/min、 27 min),离心后弃上清,沉淀称重后存于-80℃冰箱保存。蛋白自诱导表达按照文献[6]进行。
1.2.2 蛋白破碎
破碎所需的缓冲液与菌体充分混合均匀,放在冰盒上等待破碎。预冷高压破碎仪,使其达到和样品相当的温度,破碎前根据菌液按1 ∶100 加入适量的PMSF。破碎压力600 左右,破碎2 遍。破碎后取样30 μL+30 μL 上样缓冲液用来跑胶,获取结果。破碎过的菌液用大型落地离心机离心(4℃、16 000 r/min 、20 min),取上清30 μL+30 μL 上样缓冲液用来跑胶验证。离心过的上清液转移至烧杯中,等待硫酸铵沉淀。
1.2.3 硫酸铵沉淀
每毫升分别依次加入0.18 g、0.25 g、0.35 g、0.45 g 硫酸铵,用磁力搅拌器搅拌30 min,4℃离心16 000 r/min30 min,分离上清和沉淀,上清继续进行硫酸铵沉淀。每次离心后都取上清20 μL加5 μL上样缓冲液,取1 mg 沉淀加20 μL上样缓冲液等待跑胶。
1.2.4 SDS-PAGE 凝胶电泳
将制胶板进行漏检、制备8%下层胶(如表1)混匀加至胶板三分之二处、制备5%上层胶(如表1)加满胶板、插上梳子,等待30~50 min 后即可使用。进行加样、电泳、染色、脱色和观测结果。

表1 垂直电泳胶的配制
1.2.5 纯化前透析
将样品放入8-1.4 k 透析袋中,将装有蛋白的透析袋放入1.2 L 透析液的烧杯中过夜,次日取出样品到50 mL 离心管中配平离心(30 min、16 000 r/min、4℃)等待纯化。
1.2.6 蛋白纯化
1 倍柱体积纯水过柱清洗20 mL 磷酸纤维素柱料。分别用40 mL Elution buffer 和60 mL Binding buffer 过柱清洗。样品过柱后收集溜穿液,并标记流穿(FT)。接着依次使用40 mL Binding buffer、40 mL Washing buffer、40 mL Washing buffer、20 mL Elution buffer 和40 mL Elution buffer过柱,分别标记为B、W1、W2、E1 和E2。然后使用60 mL Elution buffer、40 mL Binding buffer 和100 mL 过滤纯水过柱清洗,最后用20%无水乙醇保存。将收集起来的所有样品各取200 μL 做丙酮沉淀待跑胶验证,将剩余样品置于冰上,保存于4℃冰箱[9]。
1.2.7 丙酮沉淀
200 μL样品加入2 mL 离心管中,每管中加丙酮至2 mL 并混匀。将样品放在-80℃冰箱冷冻10 min。取出样品解冻,用快速离心机14 000 r/min、4℃离心20 min。回收上清,把离心管放在干燥箱中60℃干燥10 min,干燥后取出加入20 μL上样缓冲液。
1.2.8 蛋白测浓度
用移液枪取2 μL样品蛋白加入Nano-300 微量分光光度计,通过蛋白对280 nm 波长的吸收值获得蛋白浓度,并记录。
1.2.9 蛋白浓缩
用浓缩管4 000 r/min、4℃ 浓缩至15 倍。
1.2.10 蛋白酶的活性检验
PCR 扩增验证蛋白酶的活性,5 个50 μL测试体系如表2 所示,95℃持续30 s,56℃持续20 s,72℃持续30 s;循环30,72℃延伸2 min,最终PCR 机器中维持温度在16℃。
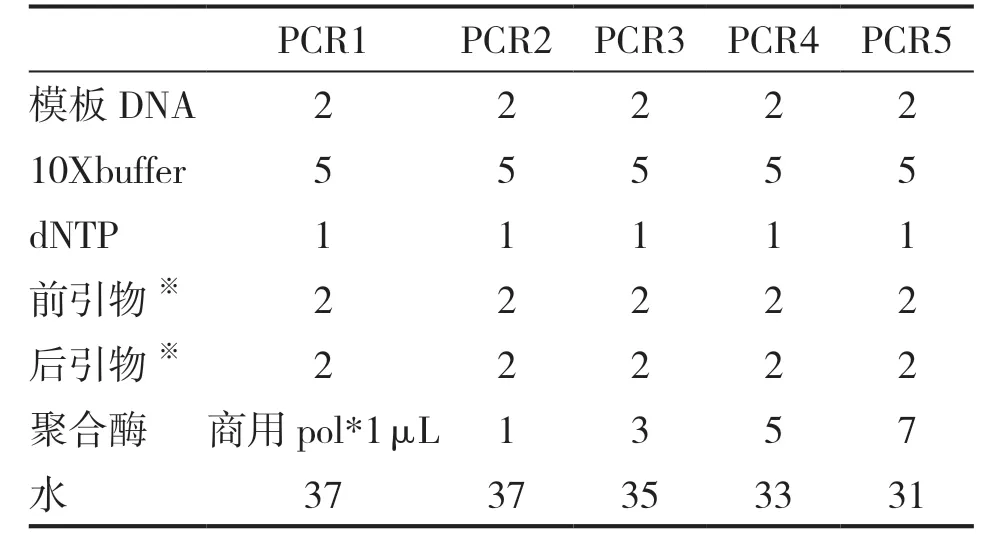
表2 目标DNA 聚合酶PCR 活性测试体系 μL
1.2.11 冷冻保存
每毫升加40%的甘油,混匀,液氮冷冻,-80℃冰箱保存得到的聚合酶。
2 实验结果
2.1 自动诱导和IPTG 诱导重组表达的比较分析
用不同的温度、时间和体积(5 mL 和10 mL)对目标蛋白进行诱导表达。结果如图1 所示,经SDS-PAGE检测可知,自动诱导28℃ 表达效果最好。

图1 诱导表达的SDS-PAGE 分析
M-7 分别是蛋白质Marker,1 和2 是诱导前样品,3 和4 是IPTG 28℃ 诱导后细胞样品,5 是IPTG 20℃ 诱导后细胞样品,6 是自动诱导25℃诱导后细胞样品,7 是自动诱导28℃ 诱导后细胞样品。星号标记处为目标蛋白条带。
2.2 硫酸铵分级沉淀蛋白
对表达的样品进行破碎后硫酸铵沉淀,硫酸铵沉淀的目的是为了达到初步纯化。结果如图2所示,经SDS-PAGE 分析可知,蛋白在0.25 g/mL和0.35 g/mL 硫酸铵沉淀时表达效果最好,因此将采用0.25 g/mL 和0.35 g/mL 沉淀进行纯化。
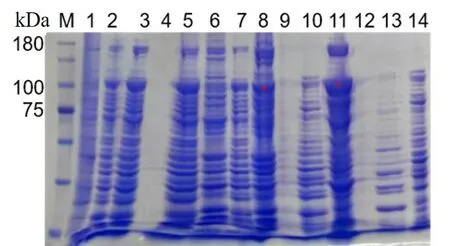
图2 破碎和硫酸铵分级沉淀SDS-PAGE 胶结果
图2 从左到右依次:M 是蛋白Marker,1 是对照,2 是破碎后蛋白,3 是破碎后蛋白上清,4是上样缓冲液,5 和6 分别是0.18 g 硫酸铵沉淀后的上清和沉淀,7 和8 分别是0.25 g 硫酸铵沉淀后的上清和沉淀,9 是上样缓冲液,10 和11 分别是0.35 g 硫酸铵沉淀后的上清和沉淀,12 是上样缓冲液,13 和14 分别是0.45 g 硫酸铵沉淀后的上清和沉淀,星号标记处为目标蛋白条带。
2.3 离子交换层析柱纯化
对硫酸铵沉淀的样品用DEAE 柱进行第一步纯化(图3a),用磷酸纤维素柱进行再次纯化(图3b)。根据纯化后丙酮沉淀SDS-PAGE 分析结果可知,W2、E1 和E2 样品含有目标蛋白,且杂蛋白含量较少。下一步将有目标蛋白的样品透析,并测蛋白活性和浓度。

图3 离子交换层析柱纯化聚合酶SDS-PAGE 胶结果
图3 (a)中M 为蛋白质Marker,1-7 分别为离心后上清、离心后沉淀、FT 样品1、FT 样品2、E1 样品、L1 样品和E2 样品;图3(b)中M 为蛋白Marker,1-6 分别为FT 样品、B1 样品、W1样品、W2 样品、E 1 样品和E2 样品。
2.4 PCR 鉴定聚合酶活性
对小规模PCR 扩增的样品跑琼脂糖凝胶电泳,用凝胶成像仪观察结果。如图4 所示,本试验表达及纯化的DNA 聚合酶比商用聚合酶的活性好,PCR 产物更多,表达更明显。
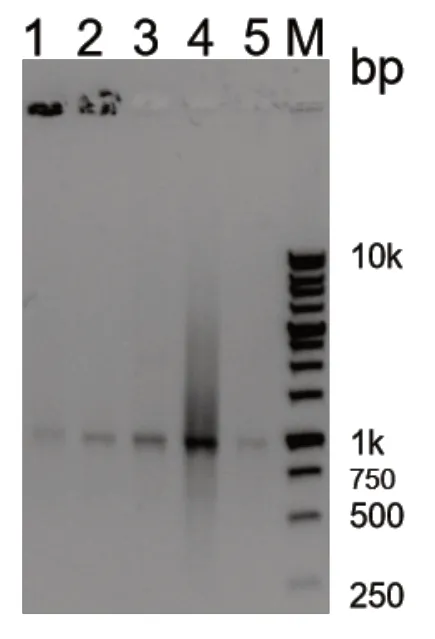
图4 PCR 测定酶的活性
M 为DNA Marker;1-4 为本试验所表达的聚合酶;5 为商用聚合酶。
3 讨论
保真度在DNA 复制中非常重要的[10]。DNA碱基配对中的错配会产生不同类型的突变,有可能导致蛋白质功能失调,并且有致癌的可能。许多DNA 聚合酶都包含1 个核酸外切酶结构域,其作用是检测碱基对是否错配,并进一步执行移除不正确的核苷酸,然后由正确的核苷酸取代。尽管不同的错配导致不同的结构特性,但DNA 聚合酶仍然能够均匀地检测和区分它们,并保持DNA复制的保真度。DNA 聚合酶还是许多诱变过程的关键,并被广泛用于生物技术中。利用大肠杆菌表达系统对目的DNA 聚合酶进行表达,有培养周期短、表达水平高、成本低等优点[11]。有关DNA 聚合酶的制备技术一直在不断的优化,主要是提高酶的表达量、特异性、酶活力等[12-13]。将DNA 聚合酶从重组大肠杆菌中提取以及纯化的方法主要有冻溶法和柱层析等方法。此外,有研究用Bio-rex70 离子交换法和聚乙二胺(PEI)沉淀对DNA 聚合酶进行纯化[14]。还有研究使用乳糖对重组大肠杆菌进行诱导,经过破胞和粗提取后,再依次过JK110 离子交换柱和SephadexG-75 凝胶柱获得纯化的聚合酶[15]。
本研究分别从菌体浓度、诱导剂浓度和诱导表达时间3 个方面进行优化得出最佳诱导条件,即 菌 液 的OD600为0.841 时 加 入1.5 mmol/L 的IPTG 进行诱导,培养时间为12 h。优化条件可使该聚合酶的大量提取生产成本降低,为大批量生产嵌合DNA 聚合酶奠定了基础。同时,通过SDS-PAGE 检验可得在此诱导条件下获取的酶纯度较高,由此可知经过本试验的纯化步骤可除去大部分杂蛋白。最后,通过PCR 扩增以及琼脂糖凝胶电泳比较商品聚合酶和本试验提取纯化的酶液的活性,结果显示两者活性相当,实验室提取纯化的嵌合式DNA 聚合酶的作用效果优于市售商品酶。
4 结论
(1)本研究测试了嵌合高保真DNA 聚合酶在不同温度和时间下的表达情况,在28℃自动诱导中蛋白表达量较高。用硫酸铵沉淀进行提纯,获得初步纯化的嵌合高保真DNA 聚合酶。
(2)采用阴离子和阳离子交换层析柱纯化后,获得较纯的嵌合高保真DNA 聚合酶。通过PCR 反应对获得高保真聚合酶进行活性分析,确认了上述工艺获得的高保真聚合酶具有聚合酶活性。
猜你喜欢
科技视界(2020年26期)2020-09-24 03:25:06
科技视界(2020年17期)2020-07-30 14:03:27
科学(2020年4期)2020-01-11 08:10:14
文艺理论研究(2018年2期)2018-06-17 10:18:06
化学教学(2018年1期)2018-02-28 21:26:29
传媒评论(2017年4期)2017-07-10 09:22:56
出版与印刷(2016年2期)2016-12-20 06:32:22
遥感信息(2015年3期)2015-12-13 07:26:52
河北省科学院学报(2014年1期)2014-07-09 02:31:18
中国药房(2012年10期)2012-08-07 03:07:18
