
基于多通道原位红外光谱技术的环氧涂层恒温固化反应研究
2023-08-08 20:41许春梁书恩蔺楠楠郝晓飞刘忠平金波孙杰睢贺良
装备环境工程 2023年7期
许春,梁书恩,蔺楠楠,郝晓飞,刘忠平,金波,孙杰,睢贺良
(1.西南科技大学 环境友好能源材料国家重点实验室,四川 绵阳 621010;2.中国工程物理研究院 化工材料研究所,四川 绵阳 621900)
由于环氧树脂具有优良的力学性能、介电性能、粘接性能以及良好的工艺适应性,使得它能够制成涂料、复合材料、浇铸料、胶粘剂、模压材料和注射成形材料[1-2],在各领域具有广泛的用途。
固化反应过程是影响环氧树脂性能的关键因素之一。目前,对于与温度相关的环氧树脂的固化反应研究,主要包括差示扫描量热法(DSC)[3-4]、动态热机械分析法(DMA)[5-6]、流变分析法[7-8]等,结合Borchardt[9]、Kissinger[10]、Freeman[11]、Doyle[12]、Ozawa[13]、Crane[10]、Coats-Redfern[11]等适用于变温速率的各种分析方法,可以用于求解环氧树脂的固化反应动力学参数。以上研究环氧树脂固化反应的表征方法中,由于不同速率条件下热效应和热惯性的作用,采用DSC 得到的活化能值相对偏高,对于固化过程和预测终点的准确性不足[14]。高聚物的支化对力学性能的影响很大,DMA 测试无法得到定量的数据,且侧基的大小以及位置的不同也会因活化能不同而在不同温度范围内出现内耗峰,导致数据不易分析。流变分析法是研究剪切应力、剪切速率和时间的关系,其研究过程中,由于剪切速率、温度、压力等流场参数的变化,使得测试过程的误差性较大。
在固化反应研究中,红外及原位红外也是多数研究者的选择[15-16],通过红外光谱分析,可以获得样品的基团结构信息。原位红外对相变和分子结构的变化敏感,可用于监测聚合物在化学反应过程中的主要特征基团吸收峰变化,有助于了解聚合物的化学反应机制及化学结构的演变,采用该方法开展固化反应动力学研究时,具有研究周期长、机时占用率高等缺点。
本文在原位红外的基础上开发了一套多通道原位反应装置,将此装置与红外光谱仪相结合,能够监测多组不同温度下样品的红外光谱,具有效率高、重复性好的特点,可弥补目前红外光谱法研究固化动力学的不足。本文以MNA 与AFG-90 的固化反应为例,通过多通道红外光谱技术,追踪了固化反应过程,希望能够以特征化学官能团的吸光度变化反映出固化度随着温度与时间的变化规律、固化方程和相应的反应动力学参数。
1 试验
1.1 材料与试样合成
试验所用材料:环氧树脂 AFG-90,环氧值为0.85 mol/100 g;甲基纳迪克酸酐(MNA),郑州阡陌进出口有限公司;乙酸乙酯,分析纯;超纯水。
室温下在烧杯中加入质量比为100∶0.2 的酸酐和水,充分搅拌得到均匀黄褐色黏稠状液体,静置24 h。然后将环氧树脂和处理后的酸酐按照1∶0.85进行混合,充分搅拌超声后,静置排除气泡,得到反应性前驱体。用胶头滴管取少量混合物于烧杯,加入适量的乙酸乙酯稀释,将稀释后的溶液均匀地滴加在KBr 盐片上,常温下待乙酸乙酯挥发后得到薄膜样品。
1.2 固化工艺与测试表征
将制备的样品放置于多通道原位反应系统的反应室中进行恒温固化。多通道原位反应装置如图1 所示。每个反应室的温度受控制台独立控制,样品放置于反应室2—6 中,而不含环氧树脂的KBr 盐片放置于反应室1 中,用于背景测试,6 个反应室的温度设置见表1。每个反应室的测量间隔为5 min,循环时间为0.5 h,固化时间为20 h。采用FTIR 记录红外光谱,每5 min 自动扫描1 次,光谱在4 000~600 cm-1内测量,分辨率为4 cm-1,每次扫描自动扣除背景,采谱总数量为240 张。

表1 反应室温度Tab.1 Reaction chamber temperature

图1 多通道原位反应装置结构[16]Fig.1 Structure of multi-channel in-situ reaction system[16]
2 结果和讨论
2.1 原位红外光谱分析
AFG-90 与MNA 的环氧树脂固化体系在30、40、50、60、70 ℃等温固化下的红外光谱如图2 所示。在图2 中,各个测试温度下的红外光谱变化相似,表明测试范围内的温度对特定结构变化或光谱吸收带确切位置的影响很小,体系的固化机制一致。其次,从酸酐的吸光度变化可知,其特征峰的强度随固化时间的增加和温度的升高变化得越明显,表明酸酐的开环速率和环氧树脂环氧基反应的速率越快。
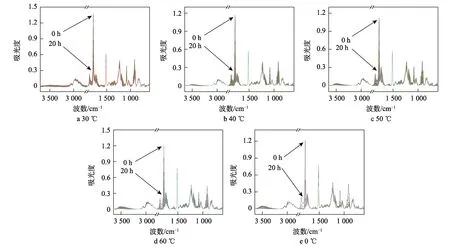
图2 环氧树脂固化体系在不同温度下的红外光谱Fig.2 Infrared spectra of epoxy resin curing system at different temperatures
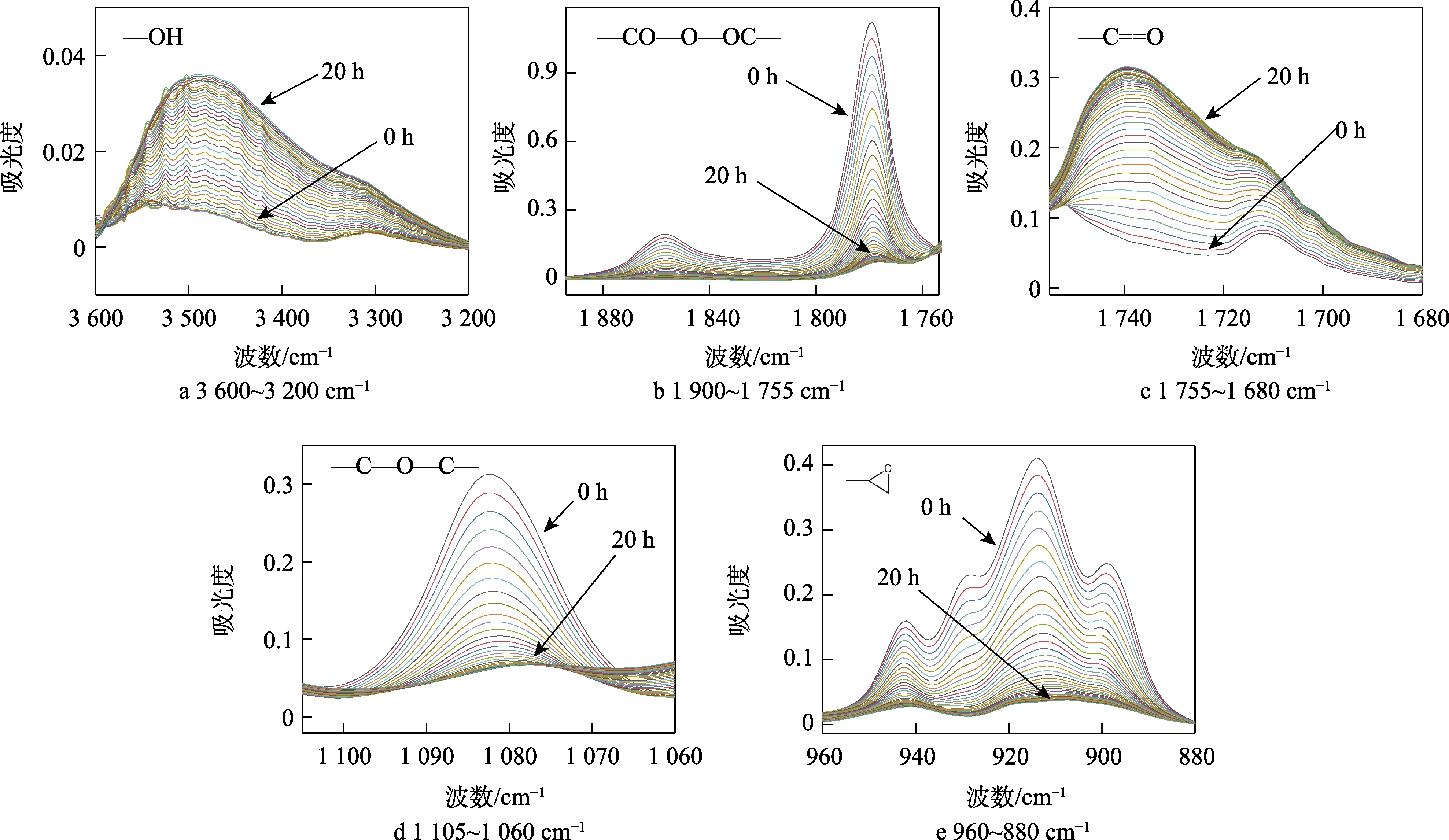
图3 环氧树脂固化体系在50 ℃下的原位红外光谱Fig.3 In-situ infrared spectroscopy of epoxy resin curing system at 50 ℃
为清楚地分析体系在固化反应中的结构变化,以50 ℃固化的红外光谱为例。在图 3 中,3 500、1 737 cm-1处分别是羟基和羰基的伸缩振动吸收峰,峰强度随固化过程不断增加。1 856、1 775 cm-1处是酸酐的伸缩振动吸收峰,而1 227、914 cm-1处归属于环氧基的伸缩振动峰吸收峰,峰强度随固化过程不断降低。这些特征峰的变化均灵敏地反映了固化反应中化学官能团的变化过程。
环氧树脂与酸酐的固化反应,通常需要体系中含有少量的醇、水、游离酸等作为促进剂,使酸酐开环形成羧酸,羧基与环氧树脂中的环氧基反应生成羟基,依次循环最终形成立体交联网络结构[17]。本试验中,体系中水的羟基以及活泼性氢使部分酸酐开环产生羧基,诱导固化反应的进行,羟基和羰基随着固化反应的进行而增加,羧基和环氧基由于发生酯化反应而减少,闭环的酸酐会借助于产生的羟基开环,羟基随链的转移使得反应依次交替进行,从而形成庞大的三维网状结构。通过以上特征官能团的变化,验证了AFG-90 与MNA 的环氧树脂体系的固化机理,如图4所示。同时也表明多通道原位红外光谱对于监测化学变化过程的有效性。

图4 环氧树脂体系的固化机理Fig.4 Curing mechanism of epoxy resin system: a) curing reaction between open-loop anhydride and epoxy resin; b) curing reaction between cyclic anhydride and epoxy resin
2.2 固化动力学
根据红外光谱中特征峰的变化,探索了环氧树脂体系固化过程中4 类主要官能团(酸酐、羰基、醚键和环氧基)的变化。在图5 中,酸酐、醚键、环氧基团的吸光度在固化过程中持续降低,羟基和羰基的吸光度在固化过程中增加。其次,反应速率随温度的升高而变快,在反应前期速率达到最高,有以下 2种原因:初始时浓度较高,使得基团之间易于发生反应;较高的温度起到诱导的作用,通过短暂的诱导形成较高的初始活性。室温下固化反应比较缓慢,且速率几乎呈现线性状态,可认为是速率相对恒定的固化过程。

图5 不同温度下的官能团吸光度与固化时间的变化Fig.5 Changes of functional group absorbance and curing time at different temperatures
通过对AFG-90 与MNA 的环氧树脂固化体系的固化反应动力学参数进行求解,并建立相应的固化动力学方程,可以有效地描述体系的固化过程,并且表观活化能Ea是固化反应能够进行所要求的最低体系能量,能够反映体系固化反应的难易程度,对固化行为的解释和固化工艺参数的确定都有着重要的指导意义。
体系在恒温状态下的固化度如图6 所示,因酸酐在1 778 cm-1波长处没有重叠峰,以其吸光度的变化作为环氧树脂固化状态的参考,固化程度可被定义为:
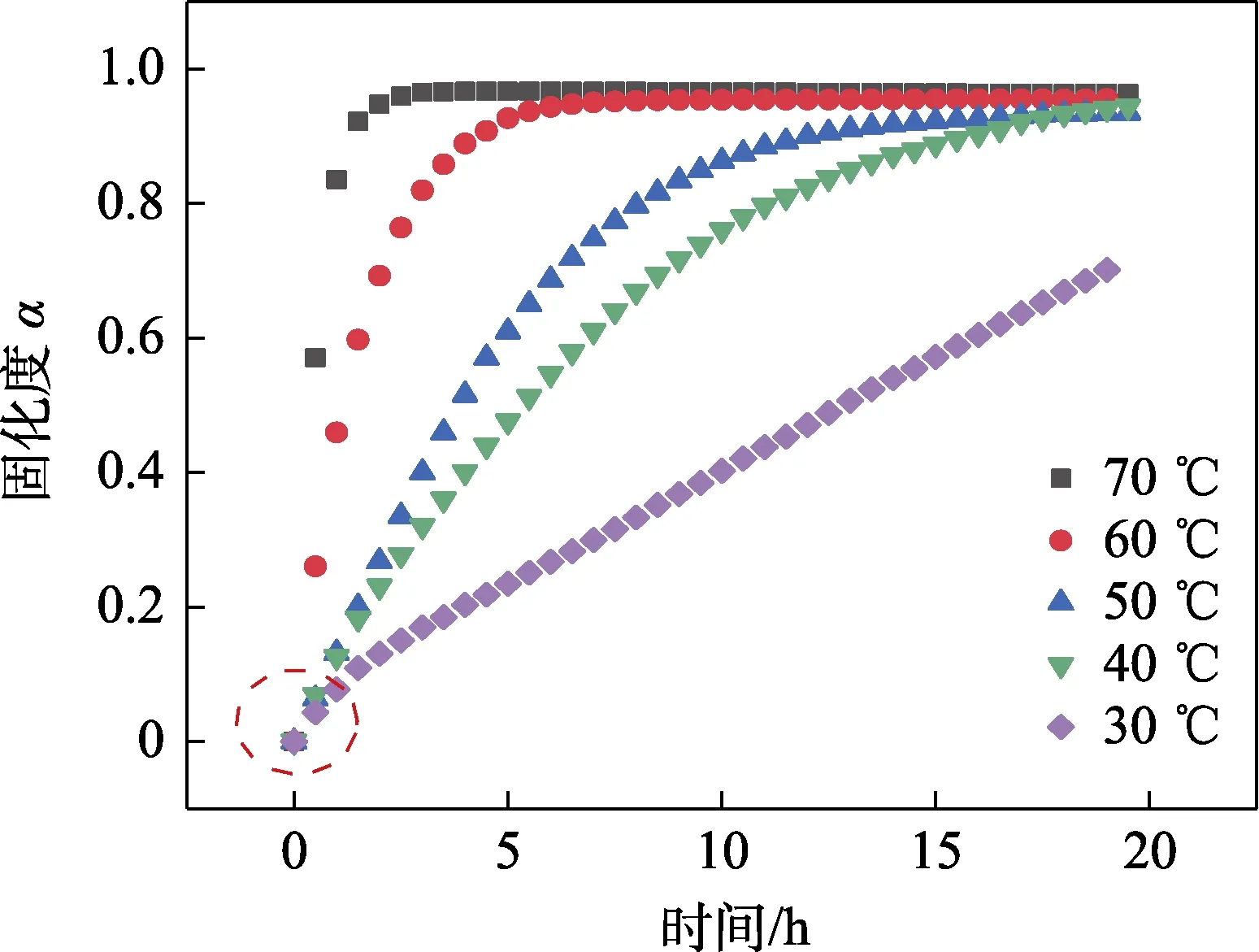
图6 环氧树脂固化体系在不同温度下的固化度Fig.6 Curing degree of epoxy resin curing system at different temperatures
式中:A0表示吸光度的初始值;At表示反应时间t时的吸光度值。
由图6 可知,不同固化温度下体系的初始转化率达到最大,即反应速率达到最快。这可能是由于固化初期反应物浓度较高,体系的黏度低,使得活性官能团之间易于碰撞相互反应。相比较低温下的转化率,较高的温度能使得体系在短时间内达到较高的固化度,之后处于恒定值,难以达到完全固化。原因可能在于,分子量短时间内变大,使得体系的黏度增加,导致分子链之间交联运动受阻。一般情况下,在固化过程中可将转化率达到10%作为固化起点,转化率达到90%作为固化结束点[18]。
一般情况下,固化反应的速率方程为[19]:
式中:α为固化度;t为反应时间;A为指前因子;Ea为表观活化能;R为气体常数;T为热力学温度;f(α)为机理函数。
当环氧树脂体系的固化度达到任意α时,通过对式(2)进行变形和积分,可得到式(3)。
当α为某一固定值时,式(3)左边是一个和温度、时间都无关的常数,通过变形可以得到式(4)。
对式(4)两边求对数,最终可得式(5)。
由式(5)可知,在某一固化度α下,确定恒定温度以及固化时间,通过以上动力学模型可求得表观活化能Ea,从而避免寻求机理的反应级数。
对图6 中每个温度下的固化度曲线进行拟合,可得不同温度下的固化方程,见表2。

表2 不同温度下的固化方程Tab.2 Curing equation at different temperatures
结合表2,计算同一温度条件下固化度α分别达到0.1、0.3、0.5、0.7、0.9 时的时间,结果见表3。

表3 不同固化度α 下的固化时间Tab.3 Curing time under different curing degrees α h
将表3 中数据代入式(5),得到lnt和1/T的关系,如图7 所示。
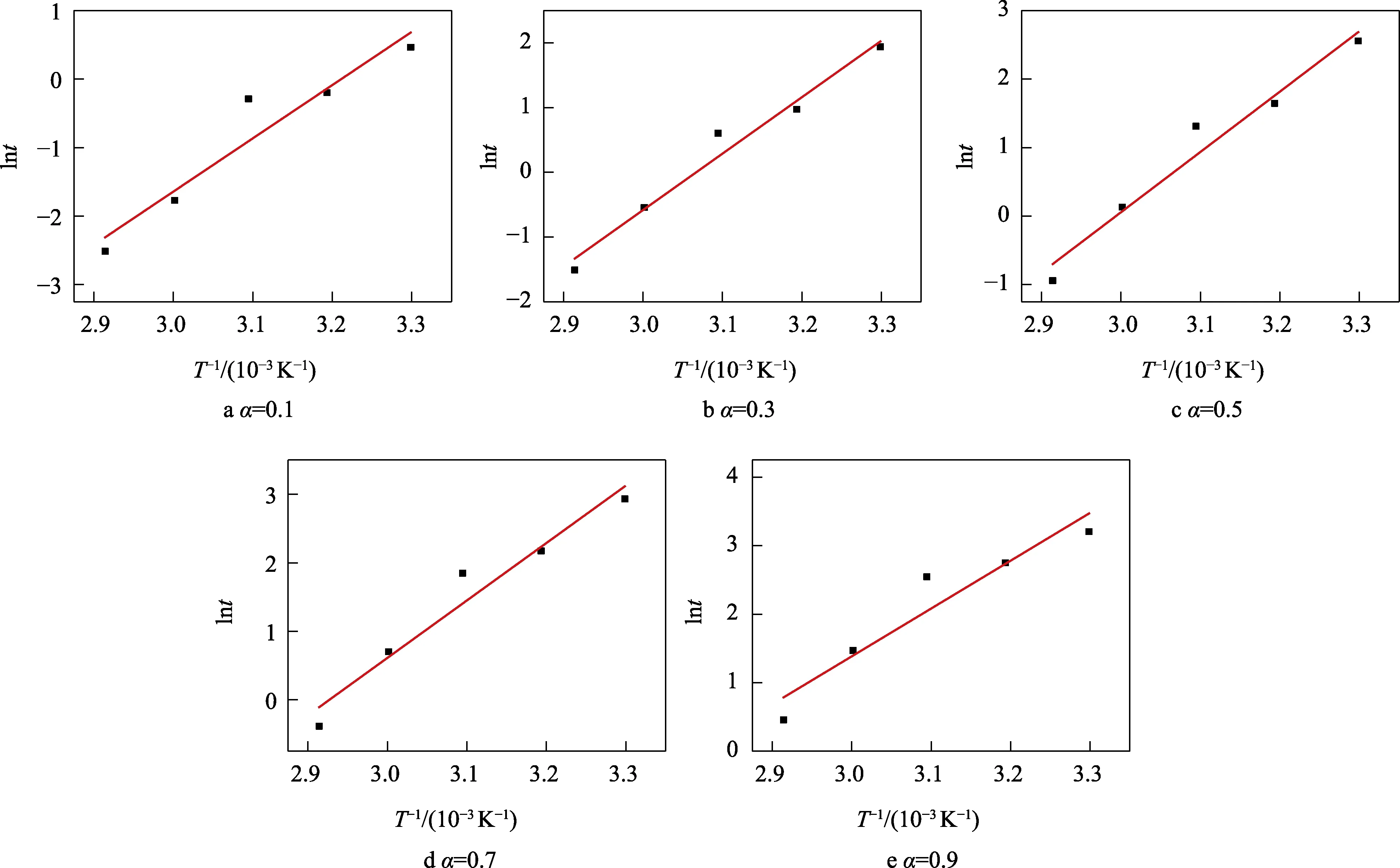
图7 不同固化度下的lnt 和1/T 之间的关系Fig.7 Relationship between lnt and 1/T under different curing degrees
通过图7 得到了不同固化程度下的动力学参数Ea的值。为得到固化过程中活化能与固化度之间更为准确的关系,将固化度为0.1~0.9 的活化能值列入表4,通过图8 展示了它们之间的关系。从图8 可以看出,环氧树脂固化体系的活化能Ea并不是恒定值,固化前期活化能较大,并且固化前期Ea值有所增加,这可能与液晶相的形成有关[20]。体系后期活化能下降,可能是固化过程由化学动力学控制向扩散控制转变所致[21-22]。整个固化过程的活化能值在 58~74 kJ/mol-1内变化,与Celina 等[23]探究所得的70~80 kJ/mol 吻合,Ea的均值为69.43 kJ/mol。

表4 不同固化度α 下的活化能Ea 值Tab.4 Activation energy Ea under different curing degrees α

图8 活化能Ea 与固化度α 的关系Fig.8 Relationship between activation energy Ea and curing degree α
3 结论
本文采用多通道原位红外光谱的表征方法,监测了AFG-90 与MNA 的环氧树脂固化体系在不同恒温下的固化过程。结合恒温动力学模型,从化学结构和表观活化能2 个方面解释了体系的固化行为,阐释了体系的固化动力学、转化率、固化状态和时间与活化能之间的函数关系。通过恒温模型的分析方法,得到了固化体系在不同固化度下的动力学参数Ea值为58~74 kJ/mol,均值为69.43 kJ/mol。因此,多通道原位红外光谱的表征方法被证明是一种有效的固化反应动力学试验方法。
猜你喜欢
大电机技术(2022年5期)2022-11-17
世界农药(2019年4期)2019-12-30
天然产物研究与开发(2018年5期)2018-06-13
材料科学与工程学报(2016年1期)2017-01-15
中国塑料(2016年2期)2016-06-15
航天制造技术(2016年6期)2016-05-09
中国塑料(2016年5期)2016-04-16
云南中医学院学报(2015年1期)2015-07-31
火炸药学报(2014年1期)2014-03-20
无机化学学报(2014年8期)2014-02-28
