
基于多源特征和双向门控循环单元的抗高血压肽识别
2023-07-28 04:20贺兴时梁芸芸
西安工程大学学报 2023年3期
贺兴时,李 锦,梁芸芸
(西安工程大学 理学院,陕西 西安 710048)
0 引 言
高血压作为最普遍的慢性疾病,影响着全球1/4以上的人口[1],高血压与心力衰竭、肾衰竭等一系列的疾病有关[2]。越来越多的人们遭受高血压的困扰,虽然有α和β受体阻滞剂、利尿剂和肾素抑制剂等新的药物,但这些药物都有血管性水肿、腹泻、皮疹等不同程度的副作用。因此,为减少或消除与高血压相关的病痛,研发更安全、副作用小,可有效抑制高血压的药物意义重大。
深度学习[3]是机器学习中的一种新兴技术,近年来已广泛应用于生物信息学的研究[4-5]。基于计算方法的抗高血压肽识别利用统计分析、数据挖掘方法提取蛋白质的序列信息,并通过机器学习算法来识别预测。WANG等提出了一种通过偏最小二乘回归方法识别蛋白质的预测模型[6]。KUMAR等设计了基于氨基酸组分、原子组成和化学描述的模型,该模型使用支持向量机对多肽进行预测[7]。WIN等提出了采用随机森林结合多种计算方法预测AHTPs 的PAAP模型[8]。MANAVALAN等构建了采用8种特征提取方法和集成分类器预测AHTPs 的mAHTPred模型[9]。ZHUANG等提出了基于预处理编码算法和卷积神经网络捕获抗AHTPs特征的模型[10]。SHI等提出一种新的AHTPs识别预测模型,该模型采用5种方法进行特征提取,合并卷积神经网络和门控循环单元(gated recurrent units,GRU)为分类器对AHTPs进行预测[11]。但这些模型存在识别精度低、过预测等缺点。
本文基于深度学习构建iAHTPs-BiGRU的AHTPs识别模型。采用多源特征提取方法从不同维度提取肽序列的信息,包括新增强分组氨基酸组分(NEGAAC)、约简的二肽组分(RDPC)、二肽频率与预期平均值之间的偏差(DDE)、氨基酸理化性质的距离变换(AAP-DT)和BLOSUM62编码,并将得到的特征信息输入到双向门控循环单元(BiGRU)中,识别蛋白质序列是否是抗高血压肽,并采用10-折交叉验证对基准数据集和独立数据集进行性能评估。
1 实 验
1.1 数据集
为了开发预测模型,方便与其他识别模型进行比较,采用构建的基准数据集和独立数据集[7]。正样本使用KUMAR等构造的抗高血压肽非冗基准余数据集[5],数据集中的所有序列均从数据库AHTPDB[12]和BIOPEP[13]得到,且都是经实验验证的正样本序列。因为较短的序列难以生成有用的信息特征,所以删除长度小于5个氨基酸残基的肽序列,剩余913条肽序列作为基准数据集的正样本。然后从Swiss-Prot中选择913条随机肽作为负样本。独立数据集是从AHTPDB和BIOPEP数据库中通过人工提取实验验证的正样本。此外,负样本仍然是从Swiss-Prot中随机肽产生。应用CD-HIT[14]删除独立数据集中与基准数据集中的序列一致度大于90%的序列,得到386个正样本和386个负样本。
1.2 特征提取方法
在基准数据集和独立数据集中,小肽、中肽和大肽等肽序列的大小不同,但是在特征提取部分,部分方法需要相同长度的肽序列。为使肽序列信息保持完整,根据最长肽序列的长度采用虚拟氨基酸“X”补齐所有序列。
1.2.1 NEGAAC方法
EGAAC将20种氨基酸分为5组[15],并已应用于病毒翻译后修饰位点预测[16]、赖氨酸琥珀酰化位点预测[17]。本文采用新的分组方法对EGAAC方法进行改进,将20种氨基酸根据亲疏水性分为6组[18],提出NEGAAC方法。新的分组为m1:R, D, E, N, Q, K, H;m2:L, I, V, A, M, F;m3:S, T, Y, W;m4:P;m5:G;m6:C。
使用滑动窗口n沿着序列进行扫描,NEGAAC计算公式为
式中:R(m,n)为基于NEGAAC的肽序列特征;W(m,n)为滑动窗口n中第m组氨基酸的数量;L为肽序列长度。通过式(1)得到(L-n+1)×6维的特征信息。
1.2.2 RDPC特征提取
RDPC是一种有效的特征提取方法,已应用于抗癌肽的识别[19]。根据氨基酸化学结构和极性将氨基酸分为r1:A, G, I, L, M, V;r2:F, W, Y;r3:H, K, R;r4:D, E;r5:C, N, P, Q, S, T等5组,即
(2)
式中:hi(i=1,2,…,25)为二肽出现的概率;ci为二肽的出现次数。
1.2.3 DDE特征表现
DDE特征表示方法[20]是将肽序列转化为数值信息,已广泛应用于蛋白质翻译后的修饰位点[21]的预测。主要步骤如下:
第一步:计算蛋白质序列二肽组分(D),计算公式为
(3)
式中:D(b,d)为氨基酸的二肽组分;wbd为氨基酸对b,d的数量。
第二步:计算肽序列的理论均值(M)和理论方差(V),计算公式为
(4)
(5)
式中:M(b,d)为肽序列的理论均值;V(b,d)为肽序列的理论方差;Ab和Ad分别为第1个氨基酸和第2个氨基酸的密码子数;Aw为氨基酸密码子的总数。
第三步:由D、M、V计算肽序列的DDE,计算公式为
(6)
式中:PDDE为基于DDE的肽序列特征。
1.2.4 AAP-DT特征方法
根据氨基酸的9种理化性质[22]将肽序列转换为数值序列。对物理化学性质的值进行标准化,基于标准化之后的数据,将每个肽序列转换为一个性质矩阵(A),即
A=(ai,j)L×9
(7)
式中:ai,j为第i个氨基酸的第j个理化性质的值。最后根据距离变换方法将理化性质矩阵转换为肽序列的特征,即
(8)
式中:PAAP-DT为基于AAP-DT方法的氨基酸特征;j1与j2为2种理化性质。根据肽序列的长度,设ε的最大值为10。
1.2.5 BLOSUM62编码
蛋白质替换计分矩阵BLOSUM是计算氨基酸之间的替换相对频率和概率,反映肽序列的进化信息,BLOSUM62由一致度大于62%的序列计算得到,已应用于原核生物赖氨酸乙酰化预测[23]、肽识别[24]等领域。基于BLOSUM62矩阵,每个氨基酸可编码为20维的特征向量。
1.3 BiGRU神经网络
为准确识别AHTPs,采用深度学习构建一个具有混合架构的神经网络。BiGRU神经网络包含输入层、BiGRU[25]、全连接层、dropout层和输出层,将数字信息矩阵输入到神经网络中。
BiGRU由前向学习和后向学习2层GRU[26]组成,故在BiGRU(记为XBiGRU)中,t时刻的隐藏状态,即
(9)

本文共设置3层BiGRU,每层的神经元个数分别为32、16和8。经过BiGRU网络得到输入矩阵的高级特征,随后将这些特征输入到全链接层进一步判别。同时为了避免过拟合,在每层的BiGRU网络中引入Dropout[27]机制,Dropout以一定的概率删除部分神经元。模型中全连接层的神经元个数分别为32、16和16,加入Dropout层提高了模型整体的泛化能力,Dropout层的参数设为0.1。全链接层为正向和反向传播,用于不断迭代更新参数和计算输出结果。采用校正线性单元[28]作为激活函数,整个网络中使用Adam[29]算法进行优化。Softmax函数以概率的形式表示分类结果,故输出层使用Softmax函数计算最终输出。
1.4 模型评估
为证明所构建的识别模型的有效性,采用10-折交叉验证方法进行性能评估并输出敏感度(Sn)、特异度(Sp)、准确度(Acc)、马修相关系数(CMC)4个评价指标,即
(10)
(11)
(12)
-1≤CMC≤1
(13)

2 结果和讨论
2.1 NEGAAC中滑动窗口n的取值
在NEGAAC中,滑动窗口n值分别取2、3、4、5、6、7、8,计算2组数据集的识别精度,不同n值的精确度如图1所示。当n=5时,基准数据集和独立数据集的识别度达到84.37%和94.65%,因此最优参数n取5。

图 1 不同n值的精确度Fig.1 Accuracy of different n value
2.2 特征提取方法
为避免所得到的特征信息过于简单,使用 NEGAAC、RDPC、 DDE、AAP-DT和BLOSUM62编码从氨基酸的理化性质和进化信息等角度表达肽序列的有效信息。为体现每个特征提取方法的识别性能,不同特征组的识别精确度结果如图2所示。

图 2 不同特征组的识别精确度Fig.2 Accuracy of different feature groups
从图2可以看出,在基准数据集中,单个特征组的识别精确度在68.63%~87.47%之间,拼接后的特征其识别准确度也达到96.78%。在独立数据集中,单个特征组的识别精确度在75.95%~96.66%之间,拼接后的特征其识别准确度达到96.78%。说明针对本文所使用的数据集,该方法识别抗高血压肽的效果相对较好,且提取的特征比单一特征识别性能更高。
对于NEGAAC特征提取方法,改进后的精确度分别达到84.37%和96.65%,比改进前分别高2.68%和2.85%,表明由NEGAAC提取出的特征更有效,更加具有识别性。
2.3 模型识别性能及不同分类器的比较
为了有效识别AHTPs,构建基于深度学习的iAHTPs-BiGRU识别模型,iAHTPs-BiGRU模型基于多源特征和深度学习对肽序列进行识别,得到了满意的结果。分类器是预测模型中的关键部分,选择不同的分类器对模型的最终输出影响较大。为选择最合适的分类器,选择9种有代表性的分类学习算法与BiGRU进行比较,其中XGboost、LGBM、ET、RF、GBDT为机器学习算法,LSTM、CNN、GRU、DNN等4种为深度学习算法,模型识别性能及不同分类器的比较见表1。
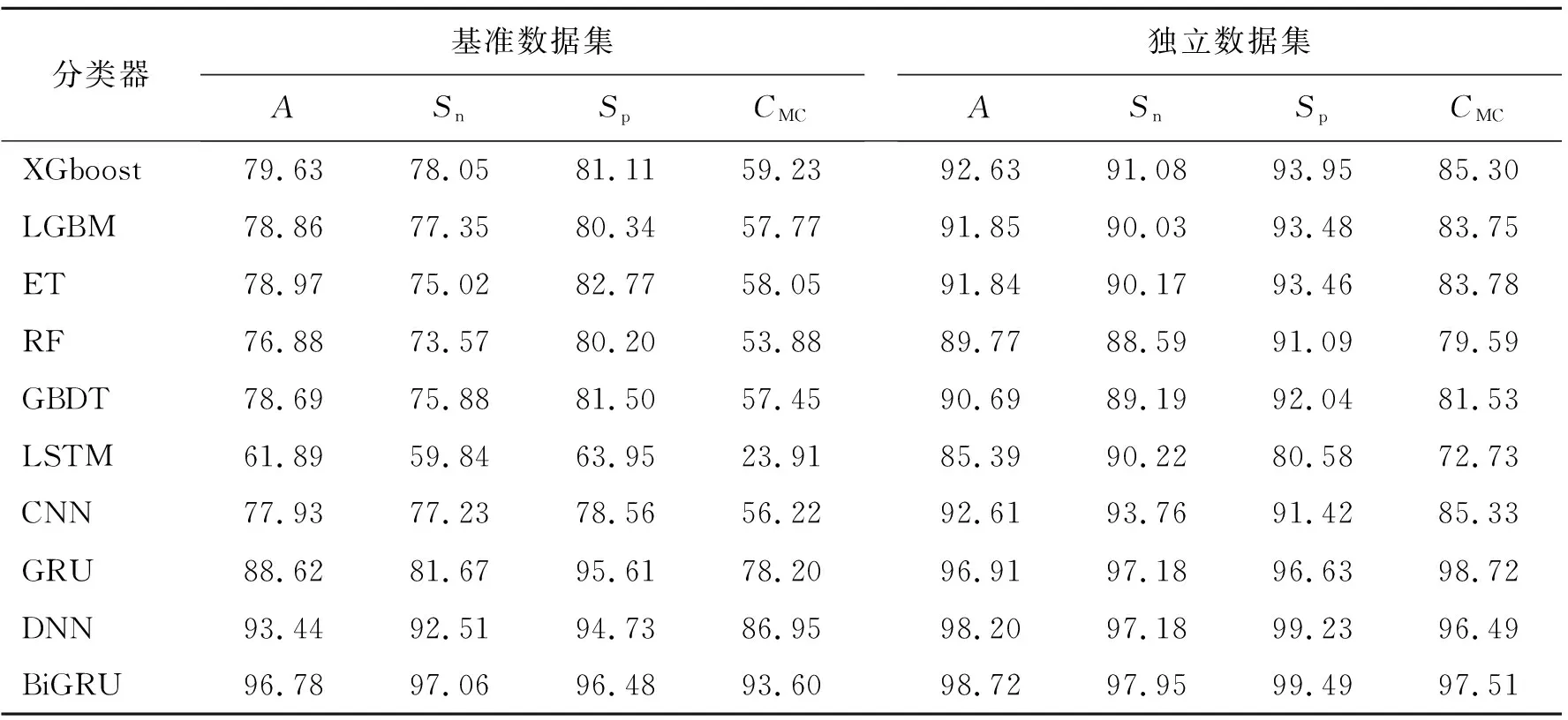
表 1 模型识别性能及不同分类器的比较
从表1可以看出,基于10-折交叉验证,在基准数据集上,评价指标A、Sn、Sp、CMC分别达到96.78%、97.06%、96.48%、93.60%。在独立数据集上,评价指标A、Sn、Sp、CMC分别达到98.72%、97.95%、99.49%、97.51%,评价指标的值都相对较高,表明iAHTPs-BiGRU模型对于识别预测AHTPs具有较好的效果。基于10-折交叉验证,BiGRU在基准数据集和独立数据集上的识别准确率分别超过其他分类器3.34%~34.89%和0.5%~13.33%,同时Sn和Sp的值相较于其他分类器也有明显的提高,进一步说明iAHTPs-BiGRU模型所使用的分类器识别性能相对较高。
2.4 识别模型性能比较
为了更加准确、公正地评价iAHTPs-BiGRU识别模型的性能,将iAHTPs-BiGRU模型性能与AHTpin_AAC[7]、 AHTpin_ATC[7]、 PAAP[8]、mAHTPred[9]、SHI等[11]5个识别模型在相同的数据集中进行比较,不同模型的精确度如图3所示。
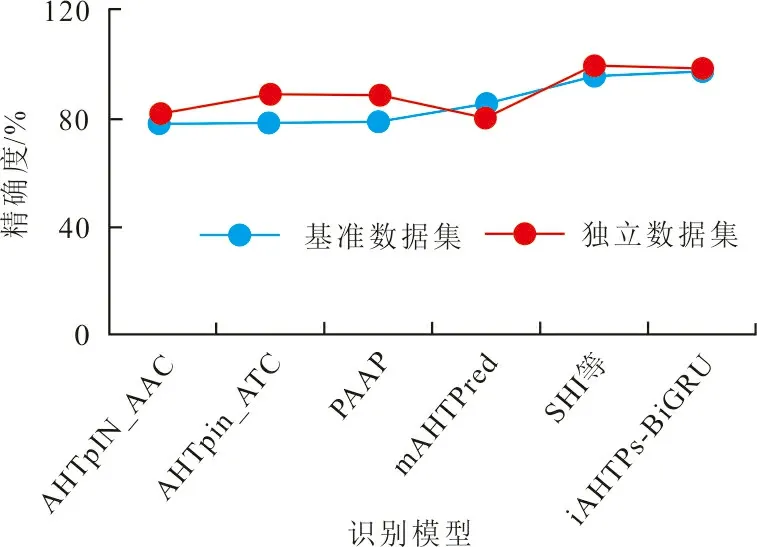
图 3 不同模型的精确度Fig.3 Accuracy comparison of different models
从图3可以看出,在基准数据集上,iAHTPs-BiGRU模型的识别准确度超出其他5个模型0.55%~18.72%;在独立数据集上,虽然比SHI等的模型识别精度低0.38%,但可以降低过拟合现象使泛化能力更强,进一步表明iAHTPs-BiGRU模型是一个有效的、可迁移性强的识别工具。
3 结 论
1) 由NEGAAC、RDPC、 DDE、AAP-DT和BLOSUM62编码等5个特征提取方法组成的多源特征信息充分表达了序列信息,提高了识别效果。
2) 基于深度学习的BiGRU算法利用多层的结构表示特征信息中的抽象特征,预测效果优良。
猜你喜欢
云南教育·中学教师(2020年11期)2021-01-07
山东煤炭科技(2020年1期)2020-03-06
电子制作(2018年19期)2018-11-14
电子测试(2018年1期)2018-04-18
自动化学报(2017年11期)2017-04-04
光学精密工程(2016年4期)2016-11-07
光学精密工程(2016年3期)2016-11-07
噪声与振动控制(2015年4期)2015-01-01
电测与仪表(2014年15期)2014-04-04
轴承(2010年2期)2010-07-28
