
淫羊藿药材中黄酮类成分提取工艺优选*
2023-07-26 07:09张秀秀王仕宝何志鹏
中国药业 2023年14期
张秀秀,杨 洁,何 洁,王仕宝△,何志鹏,房 宇,张 慧,向 仪
(1. 汉中职业技术学院药学院,陕西 汉中 723000; 2. 汉中职业技术学院秦巴山区药〈食〉用植物研究所,陕西 汉中 723000; 3. 陕西省汉中市食品药品监督检验检测中心,陕西 汉中 723000; 4. 陕西国际商贸学院医药学院,陕西 西安 712046)
淫羊藿为小檗科淫羊藿Epimedium brevicomuMaxim.、箭叶淫羊藿Epimedium sagittatum(Sieb.et Zucc.)Max⁃im.、柔毛淫羊藿Epimedium pubescensMaxim. 或朝鲜淫羊藿Epimedium koreanumNakai 的干燥叶[1],具有补肾壮阳、益精补气等功效,主治腰膝酸软、风湿痹痛、四肢麻木不仁等病症[2]。有研究表明,其在抗衰老、提高免疫力、抑制肿瘤细胞生长、控制心血管系统疾病中疗效显著[3-4]。淫羊藿总黄酮(TFE)为淫羊藿药材最主要的化学成分,现代药理学研究表明,TFE对内分泌组织、免疫系统、骨与肿瘤及心脑血管系统均有益[5-7]。基于此,本研究中通过响应面法,以朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ等6 种黄酮类成分总含量为评价指标,优选淫羊藿药材最佳提取工艺。现报道如下。
(4) 2台FEP采用自开发软件接口协议与外部时钟源同步,但无法确保2台FEP的时钟精度偏差均在50 ms内(仅采用NTP或PTP协议时才能确保精度在50 ms内),不能同时为下一层提供时钟源服务。
1 仪器与试药
1.1 仪器
1260B 型高效液相色谱仪(美国Agilent 公司);DS-8510DTH型超声波清洗器(昆山市超声仪器有限公司);XSR205DU 型精密电子天平(瑞士Mettler Toledo 公司,精度为0.01 mg)。
1.2 试药
朝藿定A1对照品(批号为P02J9S64790)、朝藿定A对照品(批号为P02J9F64787)、朝藿定B对照品(批号为G19A11L121806)、朝藿定C对照品(批号为N18GB166250)、淫羊藿苷对照品(批号为T11A11B111118)、宝藿苷Ⅰ对照品(批号为A20GB158231),均购于上海源叶生物科技有限公司(含量≥98%);乙腈、甲醇、乙醇均为色谱纯,其余试剂均为分析纯,水为超纯水。淫羊藿药材样品于2020年5月1日采集于陕西省汉中市宁强县,经汉中职业技术学院秦巴山区药(食)用植物研究所王仕宝副教授鉴定为箭叶淫羊藿的干燥叶;选取绿色鲜艳、完整叶片阴干后保存;试验前将药材样品粉碎(过3 号筛),密封,备用。
在政策和资金扶持上,各县(市)均出台了招商引资优惠政策。其中,麻江县规定对在规划区内规模连片种植3.33 hm2以上规定品种经济林的,政府均免费提供苗木或按1.35万~1.5万元/hm2的标准给予苗木费补助;对种植业主申请银行贷款用于发展经济林、中药材等水土保持林草建设的,在规定年限和贷款额度内政府给予贴息。雷山县规定利用荒山或坡耕地种植茶叶并验收合格的,一次性补助0.6万元/hm2,对每个加工厂一次性补助5万元。岑巩县政府则直接出资180万元,扶持投资者建成油茶、核桃基地667多hm2。黄平县在肥料、财税、土地、规费等方面对民间投资者给予相应扶持。
2 方法与结果
2.1 含量测定
2.1.1 色谱条件
系统适用性试验:取上述混合对照品溶液、供试品溶液各适量,按2.1.1 项下色谱条件进样测定,记录色谱图。结果,理论板数按淫羊藿苷峰计应大于8 000;分离度均大于1.5,基线分离良好。详见图1。
色谱柱:AgilentEclipsePlus-C18柱(250mm×4.6mm,5µm);流动相:水(A)-乙腈(B),梯度洗脱(0~35 min时74%A,35~36 min 时74%A →55%A,36~45 min 时55%A →53%A,45~60 min 时53%A →47%A);流速:1.0 mL/ min;检测波长:270 nm;柱温:30 ℃;进样量:10µL。
取朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ对照品各适量,精密称定,用甲醇溶解,制得质量浓度分别为675,698,684,679,657,636 µg/mL的单一对照品溶液;分别精密量取适量,混合,即得混合对照品溶液。取药材样品粉末约0.1 g,精密称定,置锥形瓶中,精密量取50%乙醇10 mL 置锥形瓶中,称定质量,超声(功率600 W、频率40 kHz,下同)提取60 min,冷却,再次称定质量,用50%乙醇补足减失的质量,摇匀,静置,取上清液,经0.22µm 微孔滤膜滤过,取续滤液,即得供试品溶液。
2.1.2 溶液制备
2.1.3 方法学考察
在1917年法国皮卡第的一场战斗中,德军突破了防线,直接冲入了英法联军的阵地,正在挖战壕的华工们猝不及防,他们只得用铁锹、镐头与德国兵肉搏,当英法联军赶到时,大部分华工已战死。
考虑到新增匝道的驶入、驶出点必须在现有道路的某一侧,因此以上图中新增匝道②-①为实例工程进行分析,可知其可行的驶入、驶出点均为3处,因此可行的新增匝道方案如图3所示。

图1 高效液相色谱图Fig.1 HPLC chromatograms
线性关系考察:分别精密量取2.1.2项下混合对照品溶液适量,制成系列混合对照品溶液,按2.1.1 项下色谱条件进样测定,记录峰面积。以待测成分质量浓度(X,µg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归。结果见表1。

表1 线性关系与检测限、定量限考察结果Tab.1 Results of the linear relation test,limit of detection and limit of quantification
提取时间:固定液料比为100,以50%乙醇分别超声提取20,30,40,50,60 min,按2.1.2 项下方法制备供试品溶液,按2.1.1 项下色谱条件进样测定,记录峰面积并计算含量。结果表明,提取40 min 时待测成分提取总量最高,随后开始逐渐下降,故选择提取时间为40 min。详见图3 b。
液料比:在液料比分别为50,100,150,200,250 的条件下,以50%乙醇超声提取60 min,按2.1.2 项下方法制备供试品溶液,按2.1.1 项下色谱条件进样测定,记录峰面积并计算含量。结果表明,液料比为50,100时待测成分提取总量较高。因液料比为50时,提取溶液过于浓稠,不方便过滤,且各成分峰面积、峰高相差较大,故选择料液比为100。详见图3 a。
稳定性试验:取2.1.2 项下供试品溶液适量,分别于室温下放置0,2,4,8,12,24 h时按2.1.1项下色谱条件进样测定,记录峰面积。结果朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ峰面积的RSD分别为2.50%,1.57%,1.92%,0.46%,1.62%,1.43%,(n= 6),表明供试品溶液在室温条件下放置24 h 内基本稳定。
重复性试验:取样品适量,精密称定,各6 份,按2.1.2 项下方法制备供试品溶液,再按2.1.1 项下色谱条件进样测定,记录峰面积。结果朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ峰面积的RSD分别为1.15%,1.83%,0.74%,1.47%,2.89%,0.92%(n=6),表明方法重复性良好。
加样回收试验:取已知含量的样品适量,共9 份,分别加入80%,100%,120%质量浓度的混合对照品溶液,按2.1.2 项下方法制备供试品溶液,再按2.1.1 项下色谱条件进样测定,记录峰面积并计算加样回收率。结果朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ的平均加样回收率分别为99.96%,100.34%,99.98%,100.48%,100.22%,101.11%,RSD分别为0.19%,0.41%,1.93%,1.95%,0.80%,2.39%(n=9)。
2.2 单因素试验
模型拟合:采用Design Expert 11 软件进行多元回归拟合,得待测成分提取总量(Y)与各因素变量的关系方程:Y= 3.118 + 0.115A- 0.034 2B+ 0.024 2C+0.055 0D- 0.192 5AB- 0.072 5AC- 0.005AD-0.01BC-0.055BD+0.06CD-0.2398A2-0.04858B2-0.098 58C2-0.187 3D2。

图2 试剂体积分数对提取总量的影响Fig.2 Effect of volume fraction of solvent on the total extraction amount
精密度试验:取2.1.2 项下混合对照品溶液适量,按2.1.1 项下色谱条件连续进样测定6 次,记录峰面积。结果朝藿定A1、朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、宝藿苷Ⅰ峰面积的RSD分别为0.38%,0.30%,0.32%,0.35%,0.56%,0.84%(n=6),表明仪器精密度良好。

图3 不同因素对提取总量的影响Fig.3 Effect of different factors on the total extraction amount
检测限与定量限考察:分别精密量取2.1.2项下混合对照品溶液适量,倍比稀释,并按2.1.1 项下色谱条件进样测定,以信噪比为3︰1、10︰1 时各成分的质量浓度分别计为检测限、定量限。结果见表1。
提取次数:固定液料比为100,以50%乙醇超声提取1~4 次,每次40 min,按2.1.2 项下方法制备供试品溶液,按2.1.1 项下色谱条件进样测定,记录峰面积并计算含量。结果表明,当提取次数超过2 次时,总黄酮的提取率缓慢下降,故选择提取次数为2 次。详见图3 c。
2.3 响应面法设计
试验设计:根据上述单因素试验结果,以乙醇体积分数(因素A)、提取时间(因素B)、提取次数(因素C)、液料比(因素D)为考察因素,待测成分提取总量为考察指标,采用响应面软件Design Expert 11.0 优选提取工艺[8-12]。因素与水平见表2,响应面试验设计及结果见表3。
1)绘图计算。主要用于图形界面的绘图操作,如绘制深度信息的时间曲线。绘图操作通过Qt的绘图类QPainter等实现。

表2 因素与水平Tab.2 Factors and their levels

表3 响应面试验设计及结果Tab.3 Design and results of the response surface test
提取溶剂:固定液料比为100(mL/ g,下同),以不同溶剂超声提取60 min,按2.1.2 项下方法制备供试品溶液,按2.1.1 项下色谱条件进样测定,记录峰面积并计算含量。结果表明,50%乙醇、50%甲醇作为提取溶剂时待测成分提取总量均较高,因甲醇毒性、挥发性较强,乙醇较安全、经济、环保,故选择50%乙醇作为提取溶剂。详见图2。
方差分析:模型回归参数方差分析结果见表4。该模型较显著(F= 4.60,P< 0.003 6),表明响应回归模型差异有统计学意义;模型中失拟项不显著(F=1.91,P =0.279 0 > 0.05),表明该模型设计合理。方程中A,AB,A²,D²对待测成分提取总量的影响有统计学意义,故可用此模型对提取工艺进行分析和预测。结果表明,因素A 具有显著意义,为主要因素;因素D 影响强度大于因素B、因素C,因素D 是影响提取总量的次要因素。
统计1970—1998年和1999—2016年Ⅰ、Ⅱ、Ⅲ区相对湿度、日平均温度、日照时间和日平均风速年序列,由图4可知,相对湿度随海拔增加而减小,海拔低于1500 m的区域相对湿度约为80%,而在1500 m以上区域,相对湿度降至60%~65%;日平均温度随海拔升高而降低;日照时间和风速则随海拔增加而增加,且日照时间随海拔变化的变化幅度和相对湿度变化较为一致。
实验结论分析 在学期末对实验组和对照组进行一对一听说测试,测试标准参照文献[5]中的“加德纳的社会—教育模式”(见图2),对幼儿进行正规语境和非正规语境两个方面的测试。本研究中正规语境的测试题目由学前教育专家指导,乡镇统一制定。测试结束后,用SPSS软件对实验组和对照组的测试成绩进行显著性差异检验。非正规语境的测试主要由监考教师反馈整理。

表4 回归模型的方差分析结果Tab.4 Results of the ANOVA of regression model
响应面预测与验证:采用Design - Expert 11.0 软件分析试验结果,得到提取总量与各因素交互作用的三维响应面图和等高线图。因素AD 间、CD 间交互作用较强,结果影响显著。淫羊藿药材最佳提取工艺为液料比为100 时,以50%乙醇提取2 次,每次40 min。详见图4。
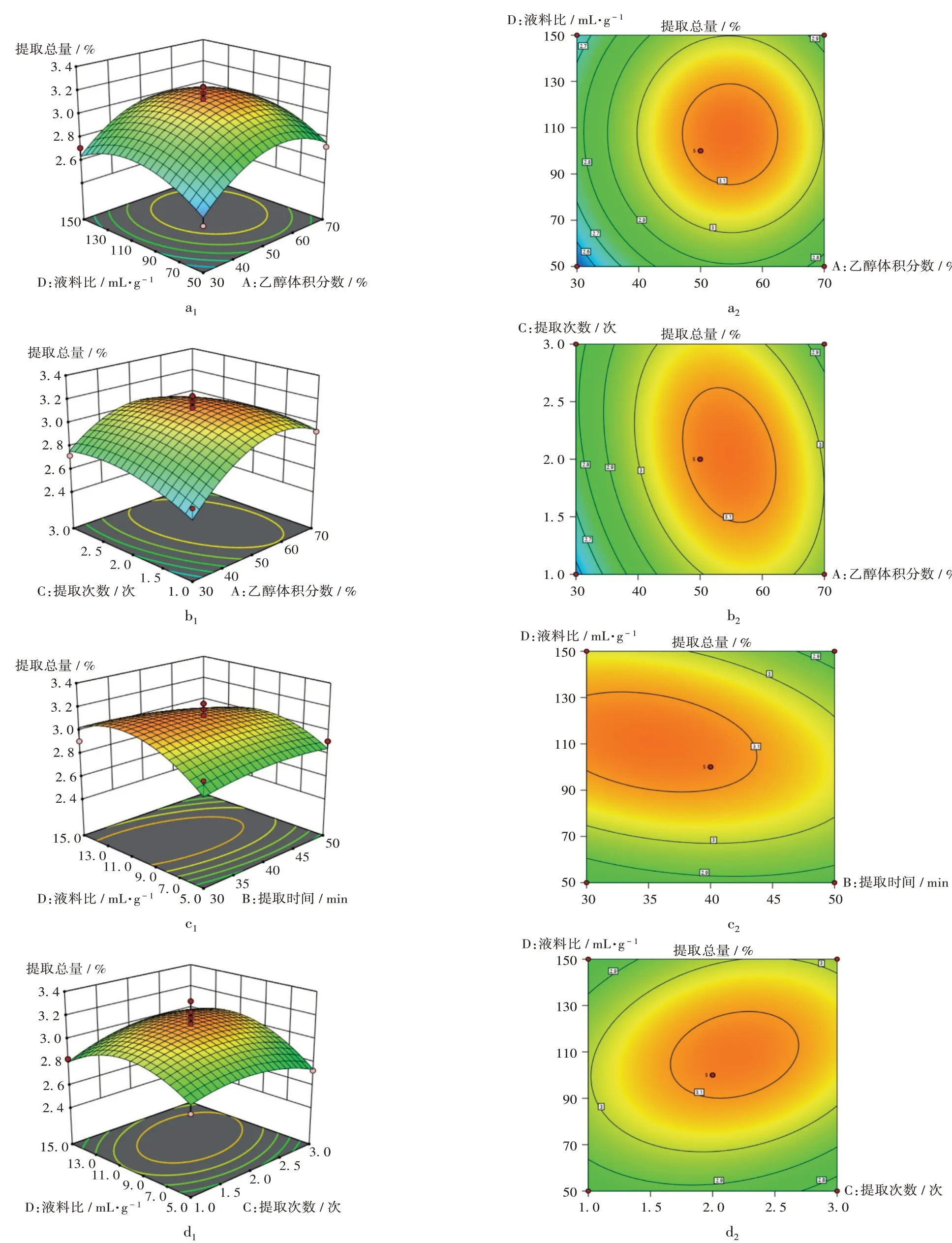
图4 不同影响因素间响应面图及等高线图Fig.4 Response surface diagrams and contour diagrams of different influencing factors
验证试验:验证最佳工艺,平行3次,结果药材样品中待测成分提取总量分别为3.16%,3.08%,3.03%,与预测值(3.12%)相当。
3 讨论
由于产地、采收季节、生长方式、干燥方式、淫羊藿药材种类、提取检测方法不同,淫羊藿药材中黄酮类成分的种类和含量也存在一定差异。预试验中考察提取工艺时,比较了醇提法、水提法、超声醇提法[13-17],发现超声醇提法对黄酮类成分的提取量相对较高,故选择该法。按响应面法软件构建的最佳提取工艺进行验证,淫羊藿叶中6种成分提取总量的实际测定值与预测值基本一致。预试验中还分别以水-乙腈、0.1%磷酸水溶液-乙腈[18]、0.1%甲酸水溶液-乙腈[19]作为流动相进行梯度洗脱,发现以0.1%磷酸水溶液-乙腈和水-乙腈作为流动相时,色谱峰峰形良好,分离度大,拖尾不明显,但由于以磷酸盐作为流动相,在质量浓度较低时易结晶析出,易造成色谱柱堵塞,冲洗色谱柱耗时较长,故选择水-乙腈作为流动相。在流动相梯度变化时间范围内,适当缩小水相的质量浓度,即可将待测6 种黄酮类成分完全分开,且出峰时间也会随之缩短,前期试验中的标准品溶液是用纯甲醇配制的,6 种成分的峰形均为前沿峰,后续在配制标准品溶液时,加入了少量的水,使其成为含水12%~15%的甲醇溶液,进样分析后发现峰形前沿情况明显改善,峰形和分离度较好。
2020 年版《中国药典(一部)》[1]明确淫羊藿苷、朝藿定A、朝藿定B、朝藿定C的总量不得少于1.5%,其中朝鲜淫羊藿不得少于0.5%。朝藿定A1因性质和朝藿定A接近,故在相同色谱条件下也能被分离出。宝霍苷Ⅰ是淫羊藿苷在体内的水解产物[2],具有显著的抗氧化、抗骨质疏松[3]、抗肿瘤活性,2020 年版《中国药典(一部)》将其列为炙淫羊藿药材含量测定的指标成分之一[1],由于淫羊藿总黄酮在淫羊藿的化学成分中的含量最高[3],药理作用强,临床应用广,故将2 种黄酮类成分纳入本次考察。
综上所述,本研究中优选的提取工艺合理可行,可为淫羊藿药材的进一步研发提供参考。
猜你喜欢
煤化工(2022年3期)2022-07-08
中成药(2018年12期)2018-12-29
科技视界(2018年22期)2018-10-08
中成药(2017年4期)2017-05-17
中国资源综合利用(2016年10期)2016-01-22
山东医药(2015年16期)2016-01-12
郑州大学学报(理学版)(2014年2期)2014-03-01
中国铸造装备与技术(2012年5期)2012-11-04
中国铸造装备与技术(2012年4期)2012-09-01
铸造设备与工艺(2010年6期)2010-11-02
