
儿童Gitelman 综合征的临床特征、基因型及预后分析
2023-07-22 05:18陈杰梅郑跃杰高晓洁南晓娟
国际医药卫生导报 2023年13期
陈杰梅 郑跃杰 高晓洁 南晓娟
1汕头大学附属深圳市儿童医院肾脏内科,深圳 518000;2汕头大学附属深圳市儿童医院呼吸内科,深圳 518000
Gitelman 综合征(Gitelman syndrome,GS)是一种罕见的常染色体隐性遗传的失盐性肾小管疾病,由编码噻嗪类利尿剂敏感的钠氯共同转运体(NCCT)蛋白的SCL12A3 基因发生突变引起钠氯重吸收障碍,导致低血钾、代谢性碱中毒、低血容量、肾素-血管紧张素-醛固酮系统(RAAS)激活等一系列相应的临床表现[1]。GS 虽属罕见病,但却是最常见的家族性低钾低镁性代谢性肾小管疾病,该病临床表现无特异性,从无临床症状到不同程度的软瘫、生长发育迟缓,甚至肾功能衰竭,与巴特综合征在临床表现存在一定的重叠,容易导致误诊和漏诊,使患儿得不到早期恰当的治疗。而严重电解质失衡可引起致死性心律失常,长期内环境紊乱可导致身材矮小、乏力等症状,严重影响患儿生活质量[2]。因此,早期诊断和治疗对GS 患儿意义重大。迄今,国内关于儿童GS 的临床表型和基因型的研究报道非常有限[3-6],目前,国内尚未有关于GS 儿童长期预后的随访报道。本研究对13 例GS 患儿的临床表现及基因表型进行回顾性分析,并对其进行长达平均4.5 年的随访,以提高临床工作者对该病的认识。
资料与方法
1.一般资料
回顾性分析2015 年1 月至2019 年12 月在汕头大学附属深圳市儿童医院住院并行基因检测明确诊断且长期随访的GS 患儿,根据GS 的定义入组GS 患儿13 例,其中男6 例、女7 例。患儿家属知情同意,且签署知情同意书;本研究经汕头大学附属深圳市儿童医院医学伦理委员会审批通过[深儿医伦审(科研)批件2022023号]。
参考《Gitelman综合征诊疗中国专家共识(2021版)》[7]确定纳入标准。(1)临床诊断标准:①存在肾性失钾及低钾血症相关临床表现,可伴有低镁血症或低钙尿症。肾性失钾:当血钾<3.0 mmol∕L 时,24 h尿钾>20 mmol;当血钾<3.5 mmol∕L时,24 h 尿钾>25 mmol。②血压正常或偏低(低于同性别同年龄同身高患儿血压P90)。③代谢性碱中毒。(2)基因诊断标准:①临床与基因完全符合:SLC12A3纯合突变或复合杂合突变;②临床与基因部分符合:SLC12A3单杂合突变[7]。
排除标准:(1)钾摄入不足或腹泻、使用利尿剂、钾分布异常等原因引起的慢性低钾血症;(2)高血压;(3)确诊年龄>18 岁;(4)未进行血气分析、血生化、尿生化、RAAS 检查;(5)未进行基因检测或基因检测未发现SLC12A3、CLCNB基因突变者;(6)未定期随访或临床资料不能获取者。
2.方法
临床资料收集。基本信息包括:性别、起病年龄、治疗前身高、血压、治疗后身高;实验室检查包括:治疗前的血pH 值、血K+、血Mg2+、血Cl-、血HCO3-、尿Ca2+、肾素、醛固酮水平,血尿蛋白尿、泌尿系超声、心电图;治疗后血K+、血Mg2+;基因检测结果。
3.统计学处理
应用SPSS 22.0 软件进行统计分析,患儿临床表型计数为计数资料,采用例(%)表示,若实验室指标为计量资料则根据是否服从正态分布采用均数±标准差(±s)或M(P25,P75)表示;二分类变量或无序多分类变量的组间比较采用χ2检验,有序多分类变量或不服从正态分布的计量资料的组间比较采用秩和检验。双侧P<0.05 表示差异有统计学意义。
结果
1.临床特点(表1)
本研究共入组13 例GS 患儿,发病年龄(6.40±3.52)年,诊断年龄(8.20±3.99)岁,最小诊断年龄为1岁11个月,最大为12岁10个月。其中0~6岁发病患儿占58%,而在0~6岁获得确诊的患儿仅占31%,仅有38%患儿在起病半年内确诊,从发病到确诊时间最长为10年;0~6岁诊断者与6岁以上诊断者的临床表现、实验室检查及基因表型差异均无统计学意义(均P>0.05)。无症状患儿占8%,其余92%患儿表现为各个系统症状。其中乏力是最常见的临床表现,出现在54%(7∕13)患儿中,23%(3∕13)患儿出现身材矮小表型,23%(3∕13)出现关节疼痛,15%(2∕13)患儿分别出现手足搐搦、烦渴嗜盐、便秘症状。所有患儿初诊时均有低钾血症,血清钾浓度为(2.60±0.37)mmol∕L,并已除外腹泻、呕吐等胃肠道丢失,10例患儿行尿钾检测,患儿的24 h尿钾均>20 mmol∕24 h,证实为肾性失钾;10 例存在低镁血症,血清镁浓度为(0.64±0.07)mmol∕L;9 例患儿存在代谢性碱中毒,11 例患儿中的5例有低钙尿症。所有患儿的肾素活性均升高,50%患儿检测出醛固酮增高。所有患儿血压均正常或偏低,均未检出QT 间期延长或甲状腺功能异常,未检测出蛋白尿、血尿、肾功能不全、钙盐沉积。
2.基因检测结果(图1~3)
在本研究13例患儿中5例患儿使用一代测序方法,2例使用多重连接探针扩增技术(MLPA),6例为全外显子检测,均检测出SLC12A3基因致病变异。其中8%(1∕13)为p.R943W纯合子,70%(9∕13)为复合杂合突变,23%(3∕13)仅发现1 个突变位点。共检出致病位点24 个,其中错义突变占71%(17∕24)、缺失突变占13%(3∕24)、无义突变占8%(2∕24)、剪切突变占4%(1∕24)、移码突变占4%(1∕24)。其中第3号染色体中T63M、第7 号染色体中T304M、第22 号外显子中R871H,这3种变异均重复出现2次。未发现CLCNB基因突变。

表1 13例Gitelman综合征患儿一般情况及临床表现比较
3.治疗及预后(图4~6)
全组13 例患儿中,均给予了补钾治疗,以氯化钾缓释片或枸橼酸钾口服为主,100%(13∕13)患儿急性期给予静脉补钾;54%(7∕13)患儿补钾同时联合螺内酯保钾治疗,15%(2∕13)患儿补钾同时联合镁剂口服补镁治疗,15%(2∕13)患儿给予吲哚美辛治疗。经治疗后所有患儿血钾、血镁均上升至3.5 mmol∕L 以上,乏力、手足搐搦等临床症状消失。患儿随访,截止至2022年6月,平均随访时长为54个月,最短33个月,最长85个月。初诊时的平均需钾量为2.63 mmol∕(kg·d),其中<6 岁起病组平均需量为2.37 mmol∕(kg·d),>6 岁组平均需钾量为2.86 mmol∕(kg·d),小年龄组钾需求量与大年龄组钾需求量差异无统计学意义(P=0.166)。随访期间,根据电解质水平及临床症状调整补钾剂量,截止2022 年4 月30 日,13 例患儿的钾需求量分别为:<6 岁起病组平均补钾需求量为6.16 mmol∕(kg·d),>6 岁组平均补钾需求量为1.48 mmol∕(kg·d),<6岁组每公斤体质量所需补钾量明显高于>6岁组(P=0.013)。13例患儿中,其中10例坚持规律补钾及定期随访,电解质水平维持在3.0 mmol∕L 以上,无自觉症状。3例患儿(病例10、病例11、病例13)10岁以后逐渐停用维持补钾药物,辅以高钾饮食,血钾波动于2.5~3.0 mmol∕L,在劳累、感染、运动量大时仍有出现乏力、烦渴、关节痛症状,在出现症状时临时口服补钾治疗。

图1 13例Gitelman综合征患儿24个致病位点的具体突变位点及方式
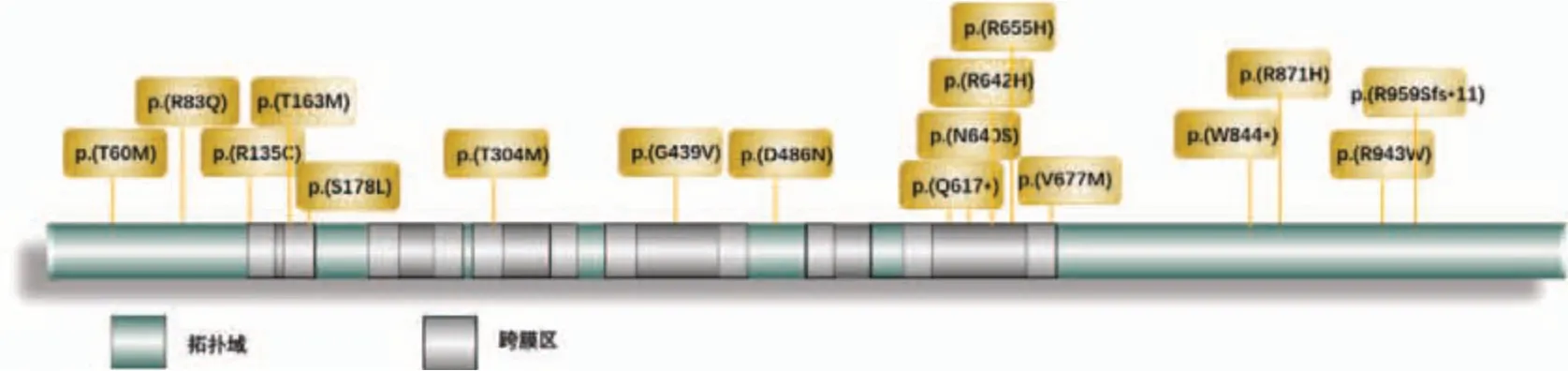
图2 蛋白质结构示意图
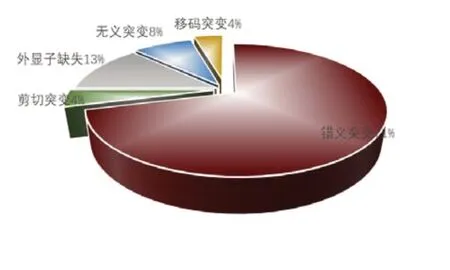
图3 13例Gitelman综合征患儿24个致病位点的SLC12A3不同变异类型的构成比
为评估患儿生长发育情况,我们对所有入组患儿初诊时的身高,随访时身高及其遗传身高进行分析,92%(12∕13)患儿的随访身高均大于其初诊时身高SDS,提示绝大部分患儿经过治疗后均出现了生长追赶。61%(8∕13)患儿随访身高SDS大于其遗传身高SDS。
讨论
1.GS的临床特点
本研究13 例患儿中,平均确诊时间为发病后(1.78±2.76)年,诊断时间从1个月到10年不等。这说明GS患儿仍因多种原因存在普遍的漏诊或误诊情况。GS 患儿常常无症状或仅有轻微症状,不易引起家长或非专科医师的重视,因此,对出现乏力、手足搐搦、恶心呕吐、烦渴嗜盐、生长迟缓等一种或多种非特异症状的患儿,可积极完善血尿生化检查,有助于早期发现可疑患者。
同为遗传性肾小管疾病,GS 常需与巴特综合征(BT)等其他低钾失盐性肾小管疾病相鉴别[8-9]。GS与BT均有低血钾、代谢性低氯性碱中毒、高肾素-血管紧张素,血压正常或偏低等类似的生化表现。GS 是编码位于肾脏远曲小管顶端膜上噻嗪类敏感的钠氯共同转运体SLC12A3基因突变引起的,远曲小管对钠重吸收功能主要起调节作用,因此,GS患者临床症状一般较BT 患者轻。BT Ⅲ型是由编码在肾小管髓袢升支粗段的氯离子通道的CLC-Kb 蛋白的CLCNKB基因突变所致,在氯离子清除试验中,对氢氯噻嗪有反应而对呋塞米无反应[10]。既往认为GS 与BT 相比,发病年龄晚、病情轻、低血镁、低钙尿、不易合并生长发育迟缓等[8-9]。但随着对GS 的认识越来越充分,越来越多早发病,病情重的GS 病例被报道,而在合并生长发育迟缓的GS 也比既往认为的多[10-11],并不是所有的GS 都存在低血镁、低尿钙等表型,交叉表型的存在让鉴别诊断存在很大的困难,在临床鉴别困难时,基因检测就显得特别重要。
2.GS的基因诊断
根据孟德尔遗传定律,常染色体隐性遗传性疾病需同一基因上的双等位基因出现变异才能致病,然而,我们的研究中发现23%患儿临床上符合GS诊断,但仅检测出1个突变位点,其他学者的研究也有同样的发现:Vargas-Poussou等[12]的448 例GS 患者的SLC12A3 基因突变的大型队列研究中,81 例患者(18%)发现1 个突变。Ma 等[13]研究结果显示,在54 例临床诊断GS 的患者中,11 例患者(20%)中发现1 个突变。Liu 等[14]对67 例中国GS 患者的研究中,发现16%的患者只携带1 个突变等位基因。Zeng 等[8]对137 例中国GS 患者的研究中发现23%的患者只携带1 个突变等位基因。这些研究均提示在该基因包括内含子及基因调控区域等非编码区域可能存在突变。而根据Vargas-Poussou等[12]的研究,在SLC12A3基因没有突变的人中,大约有1∕3的人有CLCNKB 基因突变。另外,Viering 等[15]描述了在13 个有GS 样电解质异常的家庭中发现了MT-TF 和MT-TI 的线粒体DNA(mtDNA)变异,这2 个变异分别编码苯丙氨酸和异亮氨酸的转移RNA。对患者衍生的成纤维细胞的分析显示了线粒体功能障碍,进一步的体外研究显示了NCC 活性的抑制,对于不明原因的GS 样小管病患者,应考虑对mtDNA进行遗传学调查[16]。
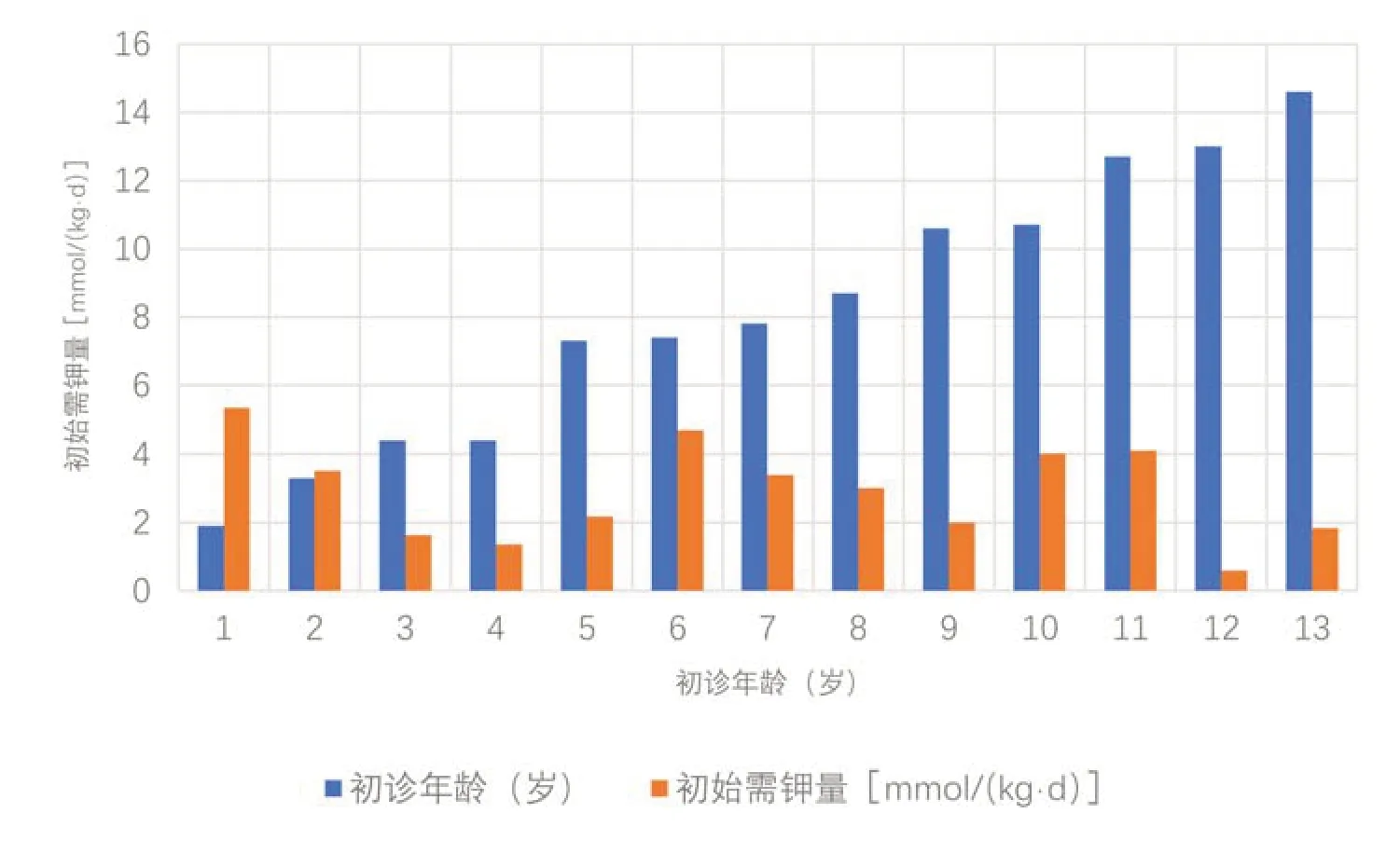
图4 13例Gitelman综合征患儿的初诊年龄与初始维持口服钾剂量
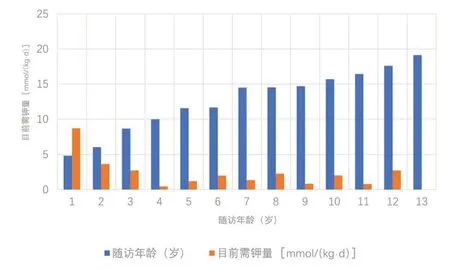
图5 13例Gitelman综合征患儿的随访年龄与目前需钾量
3.GS的治疗与预后
GS 治疗以对症治疗为主,目的是纠正电解质紊乱,缓解临床症状,减轻对生活质量的影响。首先,鼓励患者增加钠盐的摄入。其次,口服或者静脉补钾和∕或补镁。且需要个体化剂量终身补充。本研究13 例患儿中,急性期均使用静脉补钾,血钾升至正常水平后开始口服补钾维持,54%患儿联合螺内酯保钾,15%联合镁剂口服。坚持规律治疗的10例患儿血钾均维持在3.0 mmol∕L 以上,血镁在0.5 mmol∕L以上,且未再出现自觉症状。病例10、11、13在10岁后未规律治疗,血钾波动在3.0 mmol∕L 以下,均出现乏力等症状反复。从随访数据中发现,年龄越大,其公斤体质量的需钾量逐渐下降,甚至有些年长儿仅仅是高钾饮食即可维持血钾稍低于正常水平而不出现症状。但年龄小的患儿往往需要更高的额外补钾量。在本研究中,矮小症是一个不少见的临床表现,占23%,这与以往认为GS 很少出现身材发育迟缓的观点不同[10]。GS 患儿的身材发育迟缓与长期的电解质和酸碱平衡紊乱有关,经过适当的电解质补充及纠正酸碱平衡紊乱后大部分患儿可出现明显的生长追赶。早期诊断和适当的治疗是避免身材矮小的有效措施[17-18]。在本研究13例患儿中,纠正电解质水平紊乱后,除了病例4随访时身高SDS 较初诊时落后外,其余患儿均有出现生长追赶。而值得一提的是,病例4 患儿其遗传身高明显低于正常人群及其初诊时的身高SDS,这可能是造成这一结果的关键因素。如果内环境紊乱纠正后,生长速度没有明显改善的患儿,应完善生长激素(GH)激发试验来判断是否存在生长激素缺乏[19-20]。GS患儿的电解质紊乱需要个体化及终身治疗,停止用药会导致病情反复,增加严重并发症及急诊事件发生风险,并对患儿的心理状态、长期生活质量造成影响。
综上,乏力为GS患儿最常见的临床表现。GS患儿需要终身治疗,规律治疗患儿预后良好。GS 临床表现缺乏特异性,需要进行详细的实验室检查,结合基因检测,诊断及规范管理对改善患儿的长期预后及生活质量意义重大。由于本组单中心研究样本量过小,所得出的结论有待进一步验证。
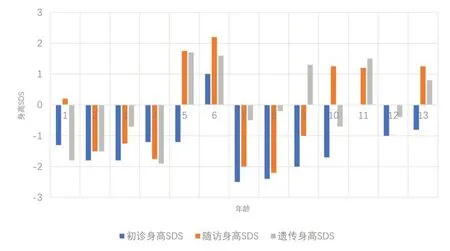
图6 13例Gitelman综合征患儿的初诊、随访、遗传身高标准差积分(SDS)
作者贡献声明陈杰梅:资料收集,数据分析,论文撰写;郑跃杰、高晓洁:构思与设计,论文修订;南晓娟:资料收集
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
中外医疗(2022年31期)2022-04-03
中医眼耳鼻喉杂志(2021年2期)2021-07-21
透析与人工器官(2020年1期)2020-11-16
儿童故事画报(2020年12期)2020-06-23
中国循环杂志(2020年4期)2020-04-27
养生大世界(2019年12期)2019-12-11
家庭科学·新健康(2018年8期)2018-10-30
猪业科学(2018年8期)2018-09-28
中国社区医师(2017年34期)2018-01-06
