
基于坏死性凋亡相关长链非编码RNA构建肝细胞癌预后模型及药物治疗反应分析
2023-07-16 03:38张玉俊朱琳赵璇地力亚尔吾斯曼江王岩
安徽医药 2023年8期
张玉俊,朱琳,赵璇,地力亚尔·吾斯曼江,王岩
作者单位:1新疆医科大学公共卫生学院,新疆维吾尔自治区 乌鲁木齐 830054;
2新疆医科大学附属肿瘤医院,新疆维吾尔自治区 乌鲁木齐 830011
肝细胞癌(hepatocellular carcinoma,HCC)是全球第三大最常见的恶性肿瘤,也是中国癌症相关死亡的第二大主要原因[1]。目前,手术切除是早期肝细胞癌的主要治疗方法。然而,由于缺乏特异性症状大多数肝细胞癌病例在诊断时已处于晚期阶段手术切除率相对较低[2]。尽管近年来在治疗方面取得了巨大进展,但由于其侵袭性的生物学特性,复发率高达70%,手术切除或肝移植成功后5 年生存率低于60%[3]。因此,确定新的诊断和预后生物标志物为HCC 的准确诊断、个体化治疗和预后预测具有重要意义。
坏死性凋亡(necroptosis)是由受体相互作用蛋白激酶RIPK1,RIPK3 和混合谱系激酶结构域样蛋白(mixed lineage kinase domain-like protein recombi⁃nant protein,MLKL)介导的程序性细胞死亡形式,其主要特征为质膜完整性的早期丧失,细胞内容物泄漏和细胞器肿胀[4]。据报道,坏死性凋亡在多种疾病的发生和发展中起着关键作用,例如神经退行性疾病,缺血性心血管疾病和癌症转移[5]。此外,坏死性凋亡通过在肿瘤微环境中释放损伤相关分子、细胞因子和趋化因子,从而产生肿瘤促进或抗肿瘤作用[6]。长链非编码RNA(long Noncoding RNA, ln⁃cRNA)是具有超过200 个核苷酸转录本的非编码RNA,在体内发挥转录调节、mRNA 剪接和mRNA 转录后调节作用[7],与肿瘤发展、转移和免疫途径密切相关,其可通过与miRNA 相互作用来调节坏死性凋亡相关基因产物,从而调节肿瘤的进程[8]。研究表明坏死性凋亡相关长链非编码RNA(necroptosis-re⁃lated long noncoding RNAs,NRLs)与多种癌症的发生发展密切相关,包括胃癌[9]、乳腺癌[10]和肺腺癌[11],而NRLs 与HCC 间的关系尚未见广泛报道。因此,进一步阐明NRLs 与 HCC 间的关系对于探索HCC新型治疗靶点和改善病人预后至关重要。
在这项研究中,通过共表达网络分析分析从TCGA 肝细胞癌(TCGA-LIHC)转录本数据中挖掘NRLs。然后,通过单变量Cox 和LASSO-Cox 回归分析,鉴定出了用于预测HCC 病人总生存期(overall survival,OS)的4 个NRLs 特征。此外,基于4 个NRLs 预后特征的药物敏感性分析为HCC 病人的临床个体化治疗提供了理论基础。
1 资料与方法
1.1 一般资料本研究所有公共数据均符合以下纳入标准:(1)纳入病人经HCC 病理确诊;(2)纳入病人的临床资料完整;(3)纳入病人有OS 的随访信息,且OS 大于30 d。从TCGA 数据库中获得374 份HCC 组织和50 份正常组织样本的RNA 测序数据和相应临床信息。根据坏死性凋亡基因集M24778.gmt和文献检索[12],共获得67 个坏死性凋亡相关基因。从GENCODE(https://www.gencodegenes.org/,截至2022 年4 月25 日)网站检索人类LncRNA 的GTF 注释文件。从以前的文献中获得47 个与免疫检查点相关的基因[11]。由于TCGA 数据是公开的,本研究不需要当地伦理委员会的批准。然而,本研究严格遵守了TCGA数据访问政策和出版指南。
1.2 方法
1.2.1 坏死性凋亡相关LncRNA 的鉴定使用从GENCODE 网站检索到的人类LncRNA 的GTF 注释文件,从TCGA-LIHC RNA-seq数据中鉴定HCC相关LncRNA。随后,通过Pearson 相关分析HCC 样本中坏死性凋亡相关基因与鉴定出的LncRNA 的共表达关系,过滤标准:(1)|Pearson 相关系数|>0.4;(2)P<0.001。为了证明NLRs 和相应的靶标mRNA 之间的相互调控连接,使用“igraph”R 包可视化lncRNAmRNA 网络。最后,采用“limma”R 包鉴定差异表达NRLs,标准为:(1)|log2 差异倍数(fold change,FC)|>1;(2)错误发现率(FDR,P值由Benjamini& Hoch⁃berg 校正进行调整)<0.05,并采用火山图进行可视化。
1.2.2 预后模型的建立与验证首先,应用单变量Cox 回归确定预后相关的NRLs(P<0.05),为防止模型的过度拟合使用 “glmnet”和“survminer”R 包进行LASSO-Cox 回归(使用10 倍交叉验证估计的惩罚参数)筛选出与HCC 预后相关的最佳NRLs,并构建预后模型。接下来,根据NRLs 的标准化表达水平和从LASSO-Cox 回归分析中得出的回归系数计算每个HCC 病人的生存风险评分。计算公式如下:风险评分=expNRL1×β1 + expNRL2×β2+ expNRL3×β3+… + expNRLn ×βn,“exp”代表已鉴定的NRL 的表达水平,“β”为通过LASSO-Cox 回归确定的系数。根据风险评分中位值将病人划分为高风险组和低风险组,使用“survival”R包分析高低风险组间OS是否存在差异(P<0.05),并采用“survminer”R 包分析临床病理特征分层高低风险组间OS 的差异(P<0.05)以了解NRLs 预后特征的适用性。随后,进行单变量-Cox 和多变量-Cox 回归分析,以探索风险评分特征是否为HCC 病人的独立预后指标(P<0.05)。最后,使用 “timeROC”“survival”R 包绘制了时间依赖的受试者操作特征(receiver operating characteristic curve,ROC)曲线以评估风险评分模型对1 年、3 年和5 年OS 的预测能力,并采用“ROCR” R 包绘制1年ROC曲线比较风险评分与传统临床特征指标的预测能力。
1.2.3 列线图的构造和校准使用“rms” R 包,建立整合临床病理学特征和坏死性凋亡相关长链非编码RNA 风险评分(NRLs risk-score)的列线图以预测HCC 病人的1 年、3 年和5 年OS。随后,开发了校准曲线来说明实际结果与预测结果之间的一致性。
1.2.4 基因集富集分析使用基因集富集分析(gene set enrichment analyses ,GSEA)软件4.1.2(http ://www.gsea-msigdb.org /gsea/index.jsp)鉴定两组中差异表达的KEGG 途径,显著富集的生物过程和途径阈值设置为P<0.05,FDR<0.25。结果由“gri⁃dExtra”“grid”和“ggplot2” R 包进行可视化。
1.2.5 肿瘤微环境分析肿瘤微环境(tumor micro⁃environment,TME)由肿瘤、基质和免疫细胞组成。基于7 种平台(XCELL、TIMER、QUANTISEQ、MCP⁃COUNTER、EPIC、CIBERSORT-ABS 和CIBERSORT)估计了TCGA-LIHC 数据集中免疫细胞的表达,并采用Spearman 相关性分析评估了免疫细胞亚群与风险评分的关系(P<0.05)。然后,使用“estimate”R 包计算TME 评分,包括基质评分(StromalScore)、免疫评分(ImmuneScore)以及总的估计评分(ESTIMATE⁃Score),并采用Wilcoxon 秩和检验比较肿瘤TIME 在高低风险组间的差异(P<0.05)。通过单样本基因集富集分析(single-sample GSEA ,ssGSEA)计算16 个免疫细胞和13 种免疫相关通路活性在高低风险组间的浸润评分差异(通过barplot 可视化)。最后,使用“ggpubr”R 包对高低风险组间的免疫检查点激活进行比较。
1.2.6 基于NRLs 特征筛选疾病潜在药物为了预测基于NRLs 特征两种不同风险群体的HCC 病人对化疗药物的反应,使用“pRRophetic”R 包来评估第8届美国癌症联合委员会(AJCC)指南强烈推荐的20种普通化疗药物的半最大抑制浓度(half-maximal inhibitory concentration,IC50)。并采用Wilcoxon 符号秩检验分析高低风险组间IC50值的差异(P<0.05)。
1.3 统计学方法本研究中使用的统计分析工具是R 软件4.0.2。采用Mann-WhitneyU检验进行高低风险组间的差异分析,运用Kaplan-Meier 方法和对数秩检验比较高低风险组病人的OS。双侧检验,P<0.05认为差异有统计学意义。
2 结果
本研究纳入来自TCGA 数据库的343 例HCC 病人以1∶1 的比例随机分配到训练集(n=172)或测试集(n=171)。研究流程如图1所示。

图1 研究流程图
2.1 鉴定HCC 病人中坏死性凋亡相关LncRNA经筛选从TCGA 数据库中共获得343 个HCC 组织和50个正常组织的样本。根据人类LncRNA的GTF注释文件,从TCGA-LIHC 基因表达文件中鉴定出16 774 个LncRNA。同时,根据坏死性凋亡基因集共鉴定出67 个坏死性凋亡基因。通过Pearson 相关分析最终获得989 个NRLs(图2A)。差异分析结果显示,760 个NRLs(9 个下调,751 个上调)在HCC 和正常组织中差异表达(图2B)。

图2 鉴定HCC病人中坏死性凋亡相关LncRNA:A为坏死性凋亡基因与 lncRNA 之间的相关网络;B为760个差异表达NRLs的火山图
2.2 模型的构建与验证在TCGA 训练集中使用单变量Cox回归分析,获得了47个与OS显着相关的NRLs(P<0.05),并制作了热图(图3A、3B)。为了避免过度拟合并提高预后特征的准确性,对这些NRLs进行了LASSO 惩罚的Cox 分析(图3C、3D),并在交叉验证误差最小点选择了4 个NRLs,使用以下公式计算风险评分:NRLs risk-Score=(0.315 1×ZFPM2-ASI)+(0.863 7×MKLN1-AS)+(0.462 1×LINC0111 6)+(1.055 6×AP003390.1)。

图3 识别HCC中NRLs预后特征:A为单变量Cox回归分析森林图;B为NRLs差异表达热图;C为NRLs的LASSO系数曲线;D为LASSO算法中变量选择的10倍交叉验证
在训练集、测试集和完整的集合中按风险评分中位值将病人划分为高风险组和低风险组,比较了HCC 病人的风险评分分布、生存状态、生存时间以及NRLs 在两组病人中的表达分布(图4A-J)。这些结果都表明高风险组病人的预后较差。同时,按年龄(Age)、性别(Gender)、分级(Grade)、分期(Stage)进行分组,研究HCC 病人在临床病理变量中的风险特征与预后的关系。结果显示,除年龄>65 岁和女性外,其余的临床病理学特征分层中低风险组的OS均显著高于高风险组,验证了风险评分模型的普遍适用性。

图4 不同集合中风险模型的预后:A~C为训练集、验证集合完整集合的风险曲线;D~F为训练集、验证集和完整集合的生存状态图;G~I为训练集、验证集和完整集合中4个NRLs表达热图
在单变量-Cox 和多变量-Cox 回归中,风险评分的风险比(HR)分别为1.06 和1.07(P<0.05)(图5A、B)。NRLs 预后特征的1 年、3 年和5 年ROC 曲线下面积(AUC)分别为0.74、0.66 和0.67(图5C)。在模型的1 年ROC 中,风险评分的AUC 为0.74,与年龄(Z=10.90,P<0.01)、性别(Z=15.25,P<0.001)、分级(Z=11.92,P<0.001)、分期(Z=14.45,P<0.001)间差异有统计学意义,表明其具有较强的预测能力(图5D)。
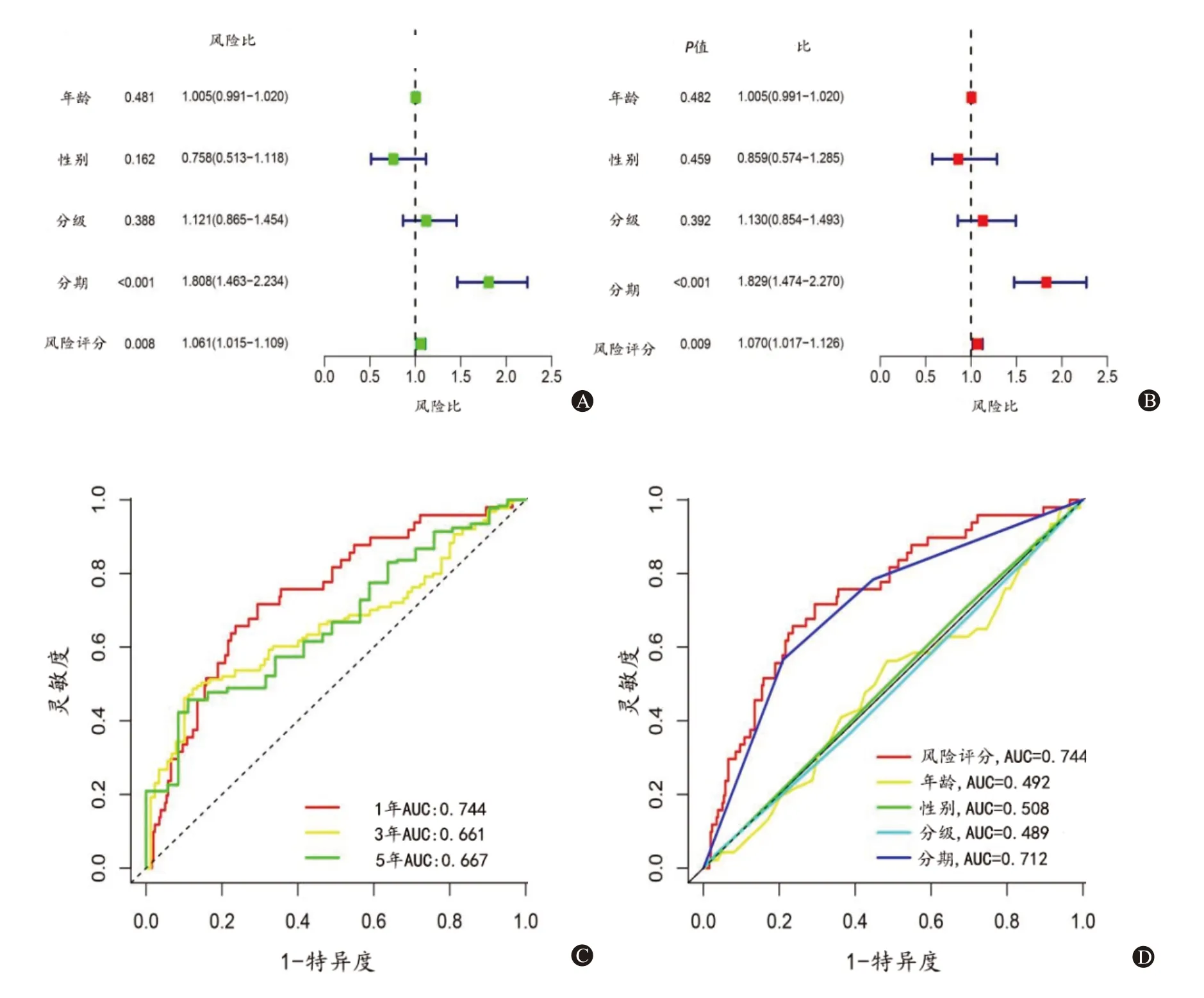
图5 验证预后特征的预测值:A为临床病理学因素和风险评分的单变量-Cox回归;B为临床病理学因素和风险评分的多变量-Cox回归;C为基于风险评分特征的1年、3年和5年ROC曲线;D为风险评分模型的预测准确性与临床病理特征的比较
2.3 列线图的构造和校准根据风险评分和临床病理学因素,开发了用于预测 1 年、3 年和5 年OS的列线图(图6A)。校准曲线显示出列线图的预测能力与实际观察值具有良好的一致性(图6B)。

图6 模型的列线图和校准曲线:A为用于预测HCC病人 1 年、3 年和 5 年OS 的列线图;B为列线图的校准曲线
2.4 GSEA 富集分析为了研究高低风险组间富集途径的差异,采用GSEA 进行KEGG 途径富集分析。结果显示,NOD 样受体信号通路、Notch 信号通路、细胞周期、RIG-I 样受体信号通路、癌症通路在高风险组中显著富集;脂肪酸代谢、初级胆汁酸代谢、药物代谢细胞色素P450、PPAR 信号通路、过氧化物酶体在低风险组中显著富集(图7)。
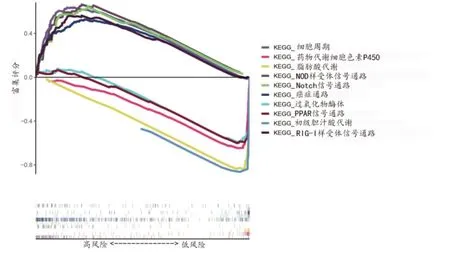
图7 高低风险组间GSEA富集分析
2.5 肿瘤微环境分析首先,我们研究了风险评分与不同平台肿瘤浸润免疫细胞间的相关性。结果显示,更多的免疫细胞与风险评分存在正相关关系(表1)。同时,免疫微环境分析结果显示,高风险病人的ImmuneScore(Z=−3.53,P<0.001)和ESTIMATE⁃Score(Z=−2.52,P<0.05)明显高于低风险病人(图8A~8C)。为了进一步探索风险评分与免疫状态之间的相关性,ssGSEA结果显示(图8D~8E),7种免疫细胞在高低风险组中存在差异包括:抗原呈递过程中像成熟树突状细胞(aDCs)、树突状细胞(DCs)、未成熟树突状细胞(iDCs)、浆细胞样树突状细胞(pDCs)、Th1 细胞(Th1 cells)、Th2 细胞(Th2 cells)、调节性T 细胞(Treg)且均在高风险组高表达。10 种免疫相关途径在高低风险组间差异有统计学意义且均在高风险组中上调,包括抗原提呈细胞共抑制(APC co inhibition)、抗原提呈细胞共刺激(APC co stimulation)、细胞因子-细胞因子受体(CCR)、检查点(check point)、人类白细胞抗原(HLA)、主要组织相容性复合体Ⅰ(MHC class Ⅰ)、副炎症(Parainfla⁃mation)、T 细胞共抑制(T cell co inhibition)、T 细胞共刺激(T cell co stimulation)、Ⅱ型干扰素反应(typeⅡ IFN Reponse)。此外,通过比较不同风险组之间的免疫检查点激活,我们发现几乎所有免疫检查点在高风险组中表达更多的活性(图8F)。

表1 不同平台免疫细胞与风险评分的相关性

图8 肿瘤微环境分析:A~C为高低风险组间 StromalScore、ImmuneScore 和 ESTIMATEScore 的箱线图;D~E为风险组中免疫细胞和免疫功能的ssGSEA评分;F为风险组间免疫检查点表达的差异
2.6 基于NRLs 特征筛选疾病潜在药物我们计算了20 种常规化疗药物在高低风险组中的IC50值,以评估HCC 病人对化疗的反应。结果显示,两个风险组对16种化疗药物的反应差异有统计学意义(P<0.05)。其中AICAR、Axitinib、Cyclopamine、DMOG、Docetaxel、Gefitinib、Lapatinib、Nilotinib、Vinblastine药物低风险组的IC50 值较低,表明这些药物对低风险组病人的疗效较好;而Cisplatin、Doxorubicin、Gemcitabine、Imatinib、Paclitaxel、Salubrinal、Tipifarn⁃ib 药物在高风险组中IC50 值较低表明对高风险组病人疗效较好。其余4个化疗药物的IC50值在高低风险组间不存在差异。
3 讨论
HCC 有较高的发病率(28.33/10 万)和死亡率(24.69/10 万)对人类健康构成极大威胁,为HCC 病人确定特异和准确的预后特征对于改善预后至关重要。先前的研究表明,坏死性凋亡在许多类型癌症的迁移和侵袭中发挥重要的作用被认为是消除癌细胞有希望的疗法[13]。lncRNA 已被证明广泛参与癌症相关细胞通路,在预后和诊断方面具有良好的预测能力[14]。在本研究中构建的风险评分模型由4个NRLs组成,根据风险评分中位值将病人划分为高低风险两组。生存分析、Cox 回归分析、ROC 曲线均表明风险评分可作为独立预后因子,构建的用于预测HCC 病人OS 的列线图显示出较好的预测性能。肿瘤微环境分析显示高低风险组间存在差异。最后,比较了高低风险组间的药物敏感性,共筛选出16种化疗药物的IC50值在两组间存在差异。
在这项研究中,获得了760 个差异表达的NRLs来探索预后功能。通过单变量Cox、LASSO 和多变量Cox 回归分析,最终确定了4 个NRLs(ZFPM2-AS1、MKLN1-AS、LINC01116、AP003390.1)与HCC病人的OS 显著相关,以构建NRLs 预后特征。ZF⁃PM2-AS1 位于染色体8q23 上,与多种癌症的发生发展密切相关。例如,肺腺癌、甲状腺癌和胃癌等[15]。研究[16]表明,ZFPM2-AS1 在HCC 组织中过表达与较差的总生存期相关,其可充当 miRNA海绵通过多个轴促进HCC细胞增殖,凋亡,迁移和侵袭。Yan等[17]在基于TCGA 的HCC 病人预后特征研究中ZFPM2-AS1 被确定为预后lncRNA。与此结果一致,本研究发现与正常组织相比HCC 组织中ZFPM2-AS1 表达显著升高。MKLN1-AS 在HCC 中可起到致癌调节剂的作用。Pan 等[18]体外细胞实验表明沉默MKLN1-AS 可抑制HCC 的HuH7 和LM3 细胞增殖、血管生成、迁移和侵袭。其可通过作为miR-654-3p的分子海绵来增加HDGF 表达,在HCC 过程中产生促癌作用,并且MKLN1-AS 的下调可抑制HCC 细胞的侵袭性表型[19]。MKLN1-AS 在HCC 中被报道可作为诊断和预后生物标志物[20],进一步证实了本研究的可靠性。LINC01116 异常表达与多种癌症相关,包括肺癌,胃癌,结直肠癌,胶质瘤和骨肉瘤[21]。其在HCC中作为癌基因起作用,参与EMT和免疫调节,过表达可促进肿瘤细胞增殖,诱导EMT 相关因子的表达,与HCC 细胞的增殖、细胞周期进展和迁移密切相关[22]。Jiang 等[23]研究表明,LINC01116 过表达的HCC 病人通常具有较低的OS,可作为HCC的预后生物标志物。AP003390.1 与HCC 关系的研究报道较少,在本研究中AP003390.1在HCC病人中高表达与病人的预后密切相关。
TME 中的免疫细胞浸润通常随着肿瘤的发生和发展而变化。在这项研究中,基于TME 评分,高风险群体中的样本具有较高的免疫评分。进一步分析表明,aDCs、DCs、iDCs、pDCs、Th1 cells、Th2 cells、Treg 均在高风险组高表达,表明这些异常浸润的免疫细胞可能与HCC 病人的肿瘤启动和发展有关。同时,APC co inhibition、APC co stimulation、CCR、Check point、HLA、MHC class Ⅰ、Parainflama⁃tion、T cell co inhibition、T cell co stimulation、Type ⅡIFN Reponse 在高风险组病人中具有较高的活性,表明HCC 的发生和发展可能与异常激活的免疫相关途径存在关联。随后,分析了常见免疫检查点表达与NRLs 预后特征之间的相关性。研究[24]指出,免疫检查点基因的表达水平与免疫疗法的疗效高度相关。结果显示,与低风险组相比,高风险HCC 病人中的大多数免疫检查点表达都升高,表明风险较高的HCC 病人接受免疫治疗时疗效更好。最后,比较了20种常见化疗药物在高低风险组中的敏感性。结果显示,9 种化疗药物对低风险组病人疗效较好,7 种化疗药物对高风险组病人疗效较好,为指导临床医生为HCC 病人选择合适的抗癌药物提供理论基础。
这项研究仍然存在一些局限性。首先,单因素-Cox 结果显示AC138356.1 是唯一对HCC 发病具有保护作用的NRLs,然而仅考虑了统计学意义将其排除在模型之外。其次,分析结果未经体内和体外验证,生物学功能需要进一步阐明。因此,在接下来的工作中将收集和扩展临床样本,并尝试通过外部实验来验证该模型的准确性。
(本文图2见插图8-7,图3,4见插图8-8,图5,6见插图8-9,图7,8见插图8-10)
猜你喜欢
核安全(2022年2期)2022-05-05
中国民间疗法(2021年18期)2021-11-02
现代畜牧科技(2021年9期)2021-10-13
昆明医科大学学报(2021年1期)2021-02-07
中学生数理化·七年级数学人教版(2019年6期)2019-06-25
中华皮肤科杂志(2018年4期)2018-01-22
China Geology(2018年3期)2018-01-13
外语教学理论与实践(2016年1期)2016-06-11
兽医导刊(2016年6期)2016-05-17
当代畜禽养殖业(2014年5期)2014-08-31
