
Molecular Dynamics Simulation of CL-20/DNDAP Cocrystal Morphology at Different Temperatures
2023-07-14 09:12:02LIXingLIWeiJUXuehai
火炸药学报 2023年6期
LI Xing, LI Wei, JU Xue-hai
(1.School of Chemistry and Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, China; 2.Third Oil Refinery, Petro China Huabei Oilfield Company, Hejian Hebei 062450, China)
Abstract:The cocrystallization of hexanitrohexaazoisowuzane (CL-20) with other energetic materials alongside the spheroidization of cocrystal morphology can reduce its sensitivity. The attachment energy (AE) model and molecular dynamics (MD) method were used to predict the morphologies of CL-20 and 2,4-dinitro-2,4-diazapentane (DNDAP) cocrystal in methyl acetate solvent at different temperatures. Meanwhile, the interaction energy between the solvent and the crystal surface was calculated to derive the attachment energy of the crystal surface, and the crystal morphologies at different temperatures were simulated. The results show that there are five morphologically dominant crystal faces of CL-20/DNDAP cocrystal under vacuum. The (1 0 0) face occupies a relative large area accounting for 37.44%, which has a significant influence on the crystal morphology. The attachment energy of CL-20/DNDAP cocrystal decreases with the increasing of temperature. The higher temperature leads to an increase in the aspect ratios, which are 1.80 and 3.93 at 280 and 360K, respectively. The lower temperature is beneficial to obtain a nearly spherical morphology. The theoretical predicted morphology of tilted prisms agrees with the experimental result.
Keywords:physical chemistry; hexanitrohexaazaisowurtzitane; CL-20; cocrystallization; 2,4-dinitro-2,4-diazapentane; crystal morphology; molecular dynamics simulation
Introduction
CL-20 is a high energy material[1]with great potential for military and civilian applications, but the problem of high susceptibility severely limits the practical applications. Some methods are needed to reduce the susceptibility without affecting the energy performance. Cocrystal is a crystal with a fixed ratio and specific structure formed by two or more components under intermolecular non-covalent bonding[2-3]. Cocrystal can change the arrangement of molecules of energetic materials to increase the energy density while reducing the sensibility and improving the safety[4-6]. There have been many studies on the ability of surface CL-20 to reduce its susceptibility by forming cocrystal crystals with other insensitive explosives. For example, 1,3-dinitrobenzene (DNB), 2,5-dinitrotoluene (DNT), benzotrifuroxan (BTF), 2-mercapto-1-methylimidazole (MMI), and 2,4-dinitro-2,4-diazapentane (DNDAP)[7-12].
The morphology of crystals is the result of a combination of internal structural factors and external conditions, and crystals exhibit different morphologies because of the differences in the growth rates of individual crystal faces. The growth rate of crystal faces is related to the type of solvent, temperature, and other factors[13-14]. Crystal morphology affects the energy and safety of energetic materials[15]. Crystals of the same size that exhibit a spherical morphology have lower impact strength and frictional susceptibility and better safety than those that exhibit a needle or plate morphology[16-20].
With the development of computer technology, computational simulation has become an important method to study the crystal morphology formation mechanism. Wang[21]found that the attachment energy of crystal faces in different solvents and the predicted crystal morphology are determined by the hydrogen bonding sites of the surfaces and are closely related to the solvent polarity through molecular dynamics simulations and modified attachment energy models. The authors gave a general mechanism to control the morphology of low-sensitivity high-energy materials with polar solvents. Song[22]simulated the effect of temperature on the crystallographic faces of 1,1-diamino-2,2-dinitroethylene (FOX-7) crystals and successfully predicted the FOX-7 crystal morphology at different temperatures, providing a new understanding of the growth and morphology of FOX-7 crystals. Cheng[23]explored the effect of solvent on the crystal morphology ofβ-HMX by simulating the crystal morphology ofβ-HMX in binary and ternary solvent systems through molecular dynamics and used radial distribution function analysis to investigate the interaction between solvent andβ-HMX. There are also many studies on the simulation of the cocrystal morphology of CL-20. Zhang[24]investigated the effect of different molar ratios of dimethyl sulfoxide-acetonitrile co-solvents on the formation of CL-20/HMX cocrystal. The results showed that the molar ratio of 1∶3 dimethyl sulfoxide/acetonitrile was favorable to the formation of CL-20/HMX cocrystal. Wang[25]simulated the formation of CL-20/ DNDA5 (2,4-dinitro-2,4-dinitropentane) at different temperatures and showed that the main driving force for the formation of CL-20/DNDA5 cocrystals is mainly the hydrogen bonding formed by H provided by CL-20 and O provided by DNDA5, and the low temperature is favorable for hydrogen bonding formation.
In this paper, molecular dynamics simulations will be used to calculate the interaction energy between each crystal face of CL-20/DNDAP cocrystal and the solvent, and the modified attachment energy model (MAE) will be combined to predict the crystal morphology of CL-20/DNDAP cocrystal at different temperatures, and analyze the effect of temperature on different crystal faces, so as to provide theoretical guidance for experimentally obtaining the spherical morphology of CL-20/DNDAP cocrystal.
1 Theory and MD Simulation Details
1.1 Theory
The attachment energy (AE) model was proposed by Hartman and Bennema. It used the Periodic Bond Chain theory[26-27]. The attachment energy (Eatt) is defined as the energy released when a crystal face with a thickness ofdhklattaches to the crystal face, and can be expressed as formula (1):
Eatt=Elatt-Eslice
(1)
whereElattis the lattice energy of the crystal;Esliceis the energy released by growing a wafer with a thickness ofdhkl.
According to the AE model[28], the relative growth rate of the crystal face (Rhkl) is positively correlated with the absolute value of the corresponding attachment energy (Eatt), see formula (2). The lower the attachment energy of the crystal face, the slower the growth rate. Slow-growing crystal face will show up in the morphology.
Rhkl∝|Eatt|
(2)
Based on the crystal structure, the growth morphology under vacuum conditions can be predicted. Important growth crystal planes are obtained. The crystal growth under solvent must overcome the resistance of the solvent layer. For this purpose, a correction of the attachment energy is required.
(3)
whereEsis an energy correction term introduced by solvent adsorption, indicating the inhibition of crystal surface growth by the solvent.Sis a correction factor reflecting the roughness of the growing crystal face, and is defined as:
(4)
whereAaccis the solvent accessible surface area of the crystal face unit (hkl).Ahklis the cross-sectional area of the crystal face unit. Obviously, the larger theSvalue, the rougher the crystal face.Escan be obtained from the interaction energy (Eint) of the solvent with the crystal face:
(5)
whereAboxis the cross-sectional area in the simulation box.Eintis the difference between the total energy (Etot) of the crystal face-solvent system, the crystal face energy (Ecry) and the solvent energy (Esol), namely:
Eint=Etot-(Ecry+Esol)
(6)
In the revised AE model, the modified adhesion energy is proportional to the growth rate of the crystal face, see equation (7):
(7)
1.2 MD simulation details
The lattice parameters of the initial cell structure of CL-20/DNDAP area=13.022Å,b=22.619Å,c=12.962Å,α=γ=90°,β=104.648°, belonging to the monoclinic crystal system. The space group isP21/c[12]. The cell structure of CL-20/DNDAP is shown in Fig.1.
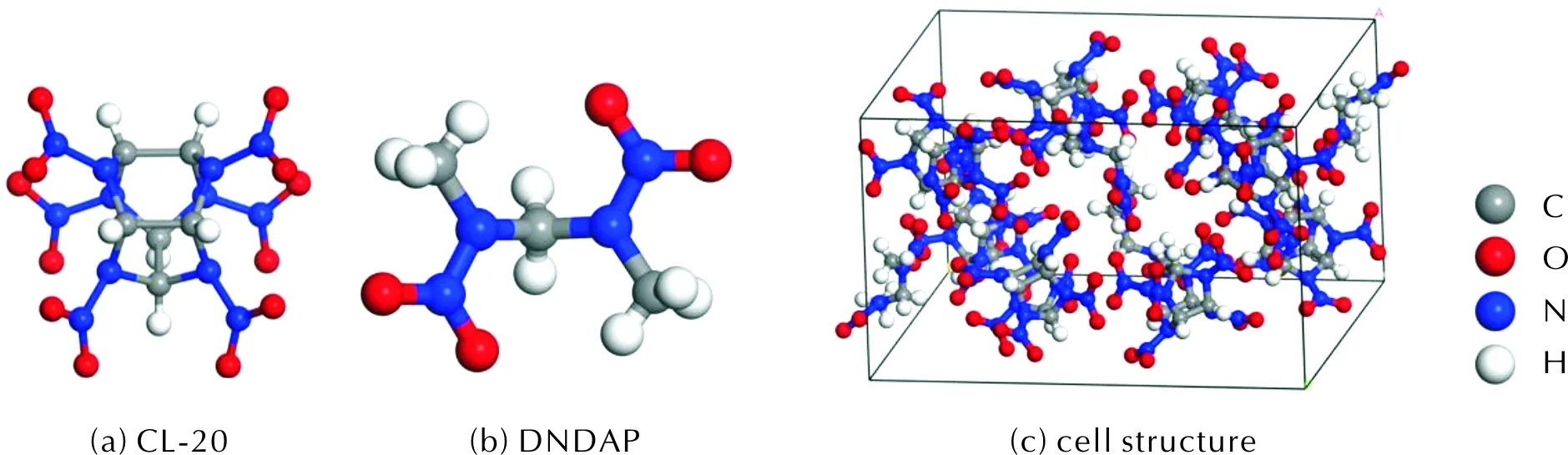
Fig.1 Molecular structures and unit cell structure of CL-20, DNDAP and CL-20/DNDAP
The COMPASS force field is the most widely used in the field of energetic materials and is suitable for the simulation of energetic compound molecules[29]. The CL-20/DNDAP unit cell structure was geometrically optimized, and the experimental lattice parameters were compared with the optimized values (Table 1). The optimized value of the crystal density at room temperature is also shown in Table 1. The relative error between the optimized lattice parameters and the experimental parameters is within 5%, indicating that the COMPASS force field is suitable for the simulation of CL-20/DNDAP.

Table 1 Comparison of experimental and optimized lattice parameters of CL-20/DNDAP
Then the AE model was used to predict the morphology of CL-20/DNDAP under vacuum, resulting in the most morphologically important growth faces. Then, the important growth crystal faces are cut, and the supercell is used to construct a periodic structure with a length and width of about 40Å. Methyl acetate was selected as the solvent system, and a solvent box composed of randomly distributed solvent molecules was constructed. The length and width of the solvent box is the same as the value of the crystal box. The number of solvent molecules is 500.
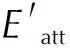
2 Results and Discussion
2.1 Crystal morphology in vacuum
The morphology of CL-20/DNDAP in vacuum was simulated with the AE model, and the main crystallographic parameters are shown in Table 2. Fig.3 shows the morphology of CL-20/DNDAP in vacuum is an angular block structure with five main growth crystallographic faces: (1 0 0), (1 1 0), (0 2 0), (0 1 1), and (1 1 -1). The morphology of CL-20/DNDAP was predicted under vacuum, and the obtained length-to-diameter ratio is 2.17, with the largest percentage of crystal face area of (1 0 0) at 37.44%. The fast-growing crystal faces disappear, while the slow-growing crystal faces appear on the final morphology. The attachment energy affects the growth rate of crystal face. The higher the attachment energy, the faster the growth rate of crystal faces and the smaller the corresponding crystal faces. On the contrary, the smaller the attachment energy of the crystal face, the larger the crystal face, and the (1 0 0) face has the smallest attachment energy and the slowest growth rate of the crystal face, so the crystal face is the largest, which is consistent with the results of other scholars[33-34].
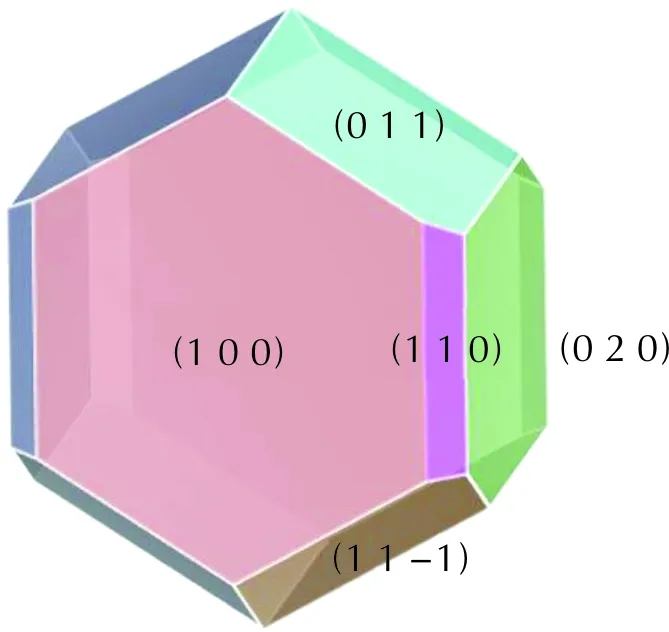
Fig.3 Crystal morphology of CL-20/DNDAP in vacuum
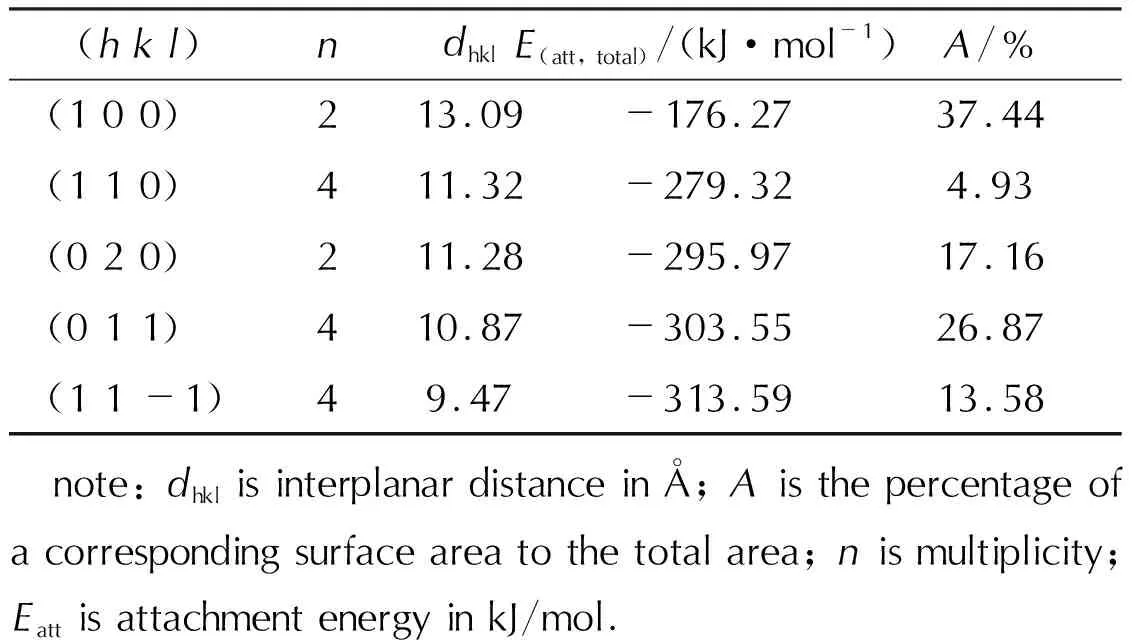
Table 2 The morphologically dominant crystal faces and related parameters of CL-20/DNDAP in vacuum
The Connolly surface of CL-20/DNDAP cocrystal is shown in Fig.4. The blue area above represents the accessible solvent surface, and it can be seen that the (1 0 0) crystal face is relatively flat, and the faces of (1 1 0), (0 2 0), (0 1 1) and (1 1 -1) have many bumps and depressions and are relatively rough, and these voids favor the adsorption of solvent molecules and thus affect the crystal face growth.

Fig.4 Connolly surfaces of each morphologically dominant crystal faces of CL-20/DNDAP
The solvent accessible area (Aacc), surface area (Ahkl) and the correspondingS(Aacc/Ahkl) values for different CL-20/DNDAP faces are presented in Table 3. The larger theSvalue, the rougher the crystal face[35], and the rough crystal face is favorable for solvent molecule adsorption. The largerSvalues for (1 1 0), (0 2 0) and (1 1 -1) crystal faces (Table 3) indicate that these faces are relatively rough and have a larger contact area with solvent molecules, and (1 0 0) crystal faces have the smallestSvalues, the flattest crystal faces, and fewer adsorption sites with solvent molecules, and the adsorption of solvent molecules with (1 0 0) crystal faces is relatively difficult. The roughness results obtained through Table 3 are consistent with the conclusions obtained in Fig.4.
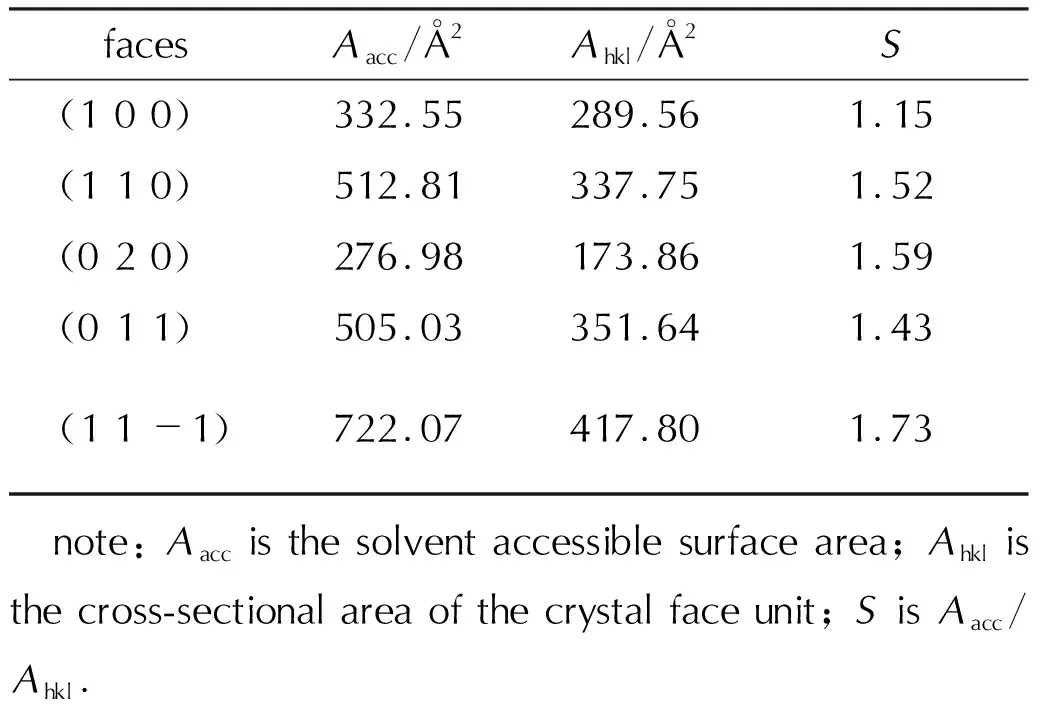
Table 3 The parameter values for the crystal habit faces of CL-20/DNDAP in solvent
2.2 Effect of temperature on morphology of CL-20/DNDAP cocrystal
Fig.5 shows the adsorption equilibrium structure of the crystal faces and the solvent after molecular dynamics calculations. Table 4 lists the CL-20/DNDAP cocrystal data calculated by molecular dynamics simulations using the modified attachment energy model with methyl acetate as the solvent and at different temperatures.

Fig.5 Adsorption equilibrium structures of the crystal faces and the solvent after molecular dynamics calculation
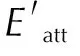
Fig.6 Comparison of the predicted crystal morphologies at different temperatures and the experimental crystal morphology at 298K from Ref[12]
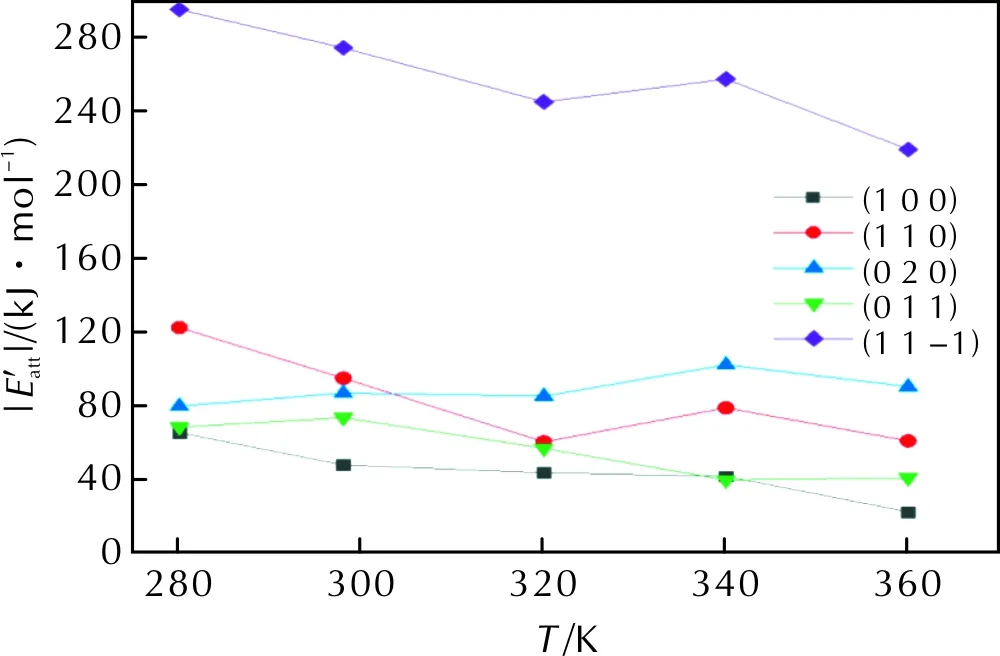
Fig.7 of each crystal face at different temperatures
By comparing the crystal morphology of CL-20/DNDAP cocrystal at different temperatures and calculating the aspect ratio, it was found that the aspect ratio increased with the increase of temperature, and the aspect ratio is related to the morphology of the crystal, and the closer the aspect ratio was to 1, the closer the crystal morphology was to spherical. From Fig.8, it can be seen that the aspect ratio is the smallest at 280K, and the aspect ratio is 1.80. This result indicates that crystallization at a lower temperature is beneficial to obtain a spherical shape.
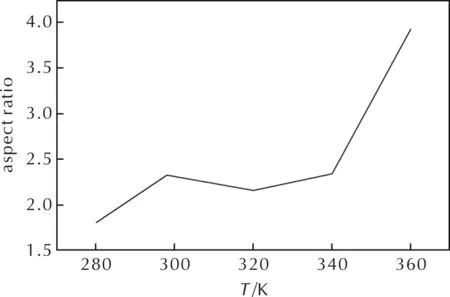
Fig.8 Predicted aspect ratio of crystal morphologies at different temperatures
3 Conclusions
(1) The simulated crystal morphologies are tilted prisms with five important growth crystal face. (1 0 0) and (0 1 1) occupy a relatively large area and are the main crystallographic faces affecting the crystal morphology.
(2) The absolute values of the attachment energy of the major crystal faces decreased with increasing temperature, indicating that the increase in temperature inhibits the growth of these crystal faces.
(3) The crystal at lower temperature is favorable to generate near-spherical morphology.
