
多信息融合和自注意力识别新冠磷酸化位点
2023-07-12 03:12:52来佳丽王明辉
重庆理工大学学报(自然科学) 2023年6期
闫 路,来佳丽,王明辉
(青岛科技大学 数理学院,山东 青岛 266042)
0 引言
2019年新型冠状病毒肺炎 (COVID-19) 是一种高度传染性的疾病,该疾病已经在世界各地迅速传播并引发了健康和社会经济危机。目前为止,还没有普遍有效的治疗方法,尽管已经有针对新型冠状病毒 (SARS-CoV-2) 感染的各种候选疫苗,但它们也可能无法控制由SARS-CoV-2引起的大流行疾病。开发有效的治疗策略来对抗SARS-CoV-2感染显得尤为紧迫。
了解SARS-CoV-2感染后宿主细胞调节的分子机制,可以确定细胞中新冠病毒蛋白的磷酸化位点,进一步推测所涉及的宿主激酶以及PTMs驱动的功能,表明它们可以在感染细胞中有效修饰。这将为新冠肺炎的药理治疗提供潜在途径[1]。然而,传统的实验方法费时费力,计算方法是更好的选择。目前,已经提出了相当多的计算方法来识别磷酸化位点。Dang等[2]开发了一种非激肽特异性磷酸化位点预测工具,使用RF作为分类器预测蛋白质磷酸化位点。MusiteDeep2017使用卷积神经网络来预测磷酸化位点[3]。DeepPSP利用深度神经网络预测磷酸化位点[4]。这些已被证明优于以前的传统机器学习方法。
目前识别新冠磷酸化位点仍然存在很多困难和挑战,主要有以下3个方面。① 不同的特征提取方法对模型的预测结果有较大的影响,而且单个特征提取方法不能很全面地提取新冠磷酸化的序列信息。② 数据冗余严重影响模型的性能,利用特征选择等方法预测结果有明显差别。③ 利用传统的分类器和一般的深度学习方法识别新冠磷酸化位点遇到了瓶颈。因此,针对以上困难,开发新的计算方法,成为了识别新冠磷酸化的研究重点。提出一种新的新冠磷酸化位点预测方法Self-DeepIPs。首先,利用4种特征提取方法,即二肽组成(dipeptide composition,DC),增强氨基酸组成(enhanced amino acid composition,EAAC),组成、转化和分布(CTD)以及BLOSUM62,将蛋白质序列信息转化为数字信息,并从多视角进行多信息融合这些特征。其次,利用互信息(mutual information,MI)去除冗余和不相关信息,最后使用构建的深度学习分类模型 (SEBI) 对新冠磷酸化位点进行分类预测,利用五折交叉验证对模型进行检验。实验结果表明,提出的Self-DeepIPs方法能够有效地鉴定识别新冠磷酸化位点。
1 材料与方法
1.1 数据来源
本研究数据集来源于Lv等[5]构建的数据集,该数据集是从文献中收集了感染SARS-CoV-2的人A549细胞的实验验证的磷酸化位点[6]。为了减少磷酸化蛋白的序列冗余并避免模型过度拟合,使用CD-HIT 程序[7],对蛋白质序列相似性大于30%的序列进行剔除。为了便于与其他现有磷酸化位点预测方法进行比较,将处理后的序列截断为33个残基长的序列片段,其中S/T位于中心。如果片段的中心S/T磷酸化,则将其定义为阳性样品,否则,它被定义为阴性样本。综上所述,得到S/T位点阳性样本5 387份,阴性样本5 387份。并将数据集随机分为严格不重叠的训练集和独立测试集,比例为8∶2。
1.2 特征提取
DC:对于20种天然氨基酸,进行两两组合可能出现400种情况,每一种两两组合的氨基酸对,称为一个二肽[8]。DC是计算给定蛋白质序列的氨基酸对的出现频率,即二肽的频率。利用DC算法,每条蛋白质序列可以生成400维特征向量。
EAAC:EAAC由Chen等[9]提出,计算固定长度子序列的氨基酸出现频率,计算公式为:
k∈(A,C,D,…,Y)
(1)
其中:win∈(window1,window2,…,windowN),N(k,win)是滑动窗口win中氨基酸类型k的个数,N(win)是滑动窗口win的大小,固定长度的序列窗口大小默认值为5。
CTD:CTD可以表征蛋白质序列的物理化学性质和氨基酸序列组成的分布模式[10]。使用13种物理化学性质来计算蛋白质特征信息,CTD可以总共可以生成273维特征向量。
BLOSUM62:BLOSUM62矩阵[11]建立在氨基酸序列的比对上,2个肽序列之间的同一性不超过62%。BLOSUM62矩阵中的每一行都被用来编码20个氨基酸之一,序列长度为n的氨基酸序列可得到20×n维的特征向量。
1.3 特征选择
MI[12]可以度量统计量之间存在的关联性,捕捉变量之间的线性和非线性关联。假设2个离散随机变量S和T的MI定义为:
(2)
其中:p(·)为概率函数。根据定义,(i)MI(S,T)=MI(T,S),(ii)MI(S,T)≥0,独立随机变量相等。
1.4 深度学习框架
本研究构建的深度网络框架由自注意力机制[13]、双向长短时记忆网络(BILSTM)[14]和全连接层结合构建,称为SEBI。构建的深度学习网络框架融合了多个组件的优点,可以使学习更有效。接下来,将按顺序介绍框架中从输入到输出的所有组件。
1) 自注意力机制
为了更好地捕捉蛋白质序列之间的交互信息,在模型中采用了一种自注意机制[13]。自注意力机制可以捕获蛋白质序列中上下信息之间的长期依赖关系。它可以有选择性地对一些重要蛋白质信息给予更多的关注,给予较高的权重,而对其他信息给予较低的权重。首先,根据上一层输出的嵌入向量得到Q、K、V3个注意力向量表示:
Q=WQX,K=WKX,V=WVX
(3)
其中:矩阵Q、K、V分别表示一组查询、键和值(输入/输出序列),WQ、WK、WV表示学习的线性运算,对Q和K进行缩放点积运算,得到相似度权重,然后利用softmax函数对相似度权重进行归一化。注意力矩阵T的计算如下:
(4)
其中:dk是一个比例因子;softmax是一个列式归一化函数;T表示注意力矩阵。
2) BILSTM
BILSTM[14]的工作原理类似于RNN。然而,除了称为细胞的内部处理单元之外,它们在称为遗忘门、更新门和输出门的循环神经元的门的使用上有所不同。在输入层的顶部使用2层BILSTM。BILSTM在每个序列索引位置l的门一般包括遗忘门、输入门和输出门3种。遗忘门可以表示为:
fl=σ(Wfhl-1+Ufxl+bf)
(5)
输入门由2部分组成,第一部分使用了ReLU激活函数,输出为il,第二部分使用了tanh激活函数,输出为al,两者的结果相乘去更新细胞状态,可以表示为:
il=σ(Wihl-1+Uixl+bi)
(6)
al=tanh(Wahl-1+Uaxl+ba)
(7)
其中:Wi、Ui、Wa和Ua为线性相关系数,bi和ba为偏置向量,σ为ReLU激活函数。细胞状态Cl由2部分组成,其公式为:
Cl=Cl-1⊗fl+il⊗al
(8)
隐藏状态hl的更新由2部分组成,即
ol=σ(Wohl-1+U0xl+bo)
(9)
hl=ol⊗tanh(Cl)
(10)
(11)
随后,将上面得到的隐藏状态输入到全连接神经网络并利用式(11)将结果输出。
Yt=htWhq+bq
(12)
本研究的模型使用Dropout防止模型过拟合,ReLU作为模型的激活函数,使用Adam函数计算每个参数的自适应学习率,categorical_crossentropy作为交叉熵损失函数来评价模型的优劣,softmax 用于对新冠磷酸化位点和非新冠磷酸化位点行分类,整个深度学习框架通过TensorFlow实现。
1.5 模型评估
为了有效地评估模型的性能,选择五折交叉验证和独立测试集对模型进行性能评估,选用准确性(ACC)、特异性(Sp)、敏感性(Sn)与马氏关系数(MCC)来作为评估模型性能的指标,具体公式如下:
(13)
(14)
(15)
(16)
其中:TP表示正确预测新冠磷酸化位点的数量,定义为真阳性;TN表示正确预测非新冠磷酸化位点的数量,定义为真阴性;FP表示错误的预测为新冠磷酸化位点的数量,定义为假阳性;FN表示错误预测非新冠磷酸化位点的数量,定义为假阴性。此外,还选择ROC和PR 曲线[15]作为衡量模型优劣的标准,AUC和AUPR分别是ROC和PR曲线下面积,曲线下面积越大代表模型的泛化能力和鲁棒性越好。统计性检验也通常被用来作为验证预测模型是否有效的方法,双侧T检验方法可以用来评估交叉验证的显著性是否明显。采用双侧T检验检测模型的。
为方便起见,提出的新冠磷酸化位点预测方法称之为Self-DeepIPs,所提出的模型的总体架构流程如图1所示。实验环境为:Windows Server 2012R2 Intel (R) Xeon (TM) CPU E5-2650@2.30 GHz with 32.0 GB of RAM,implemented by Python 3.7。
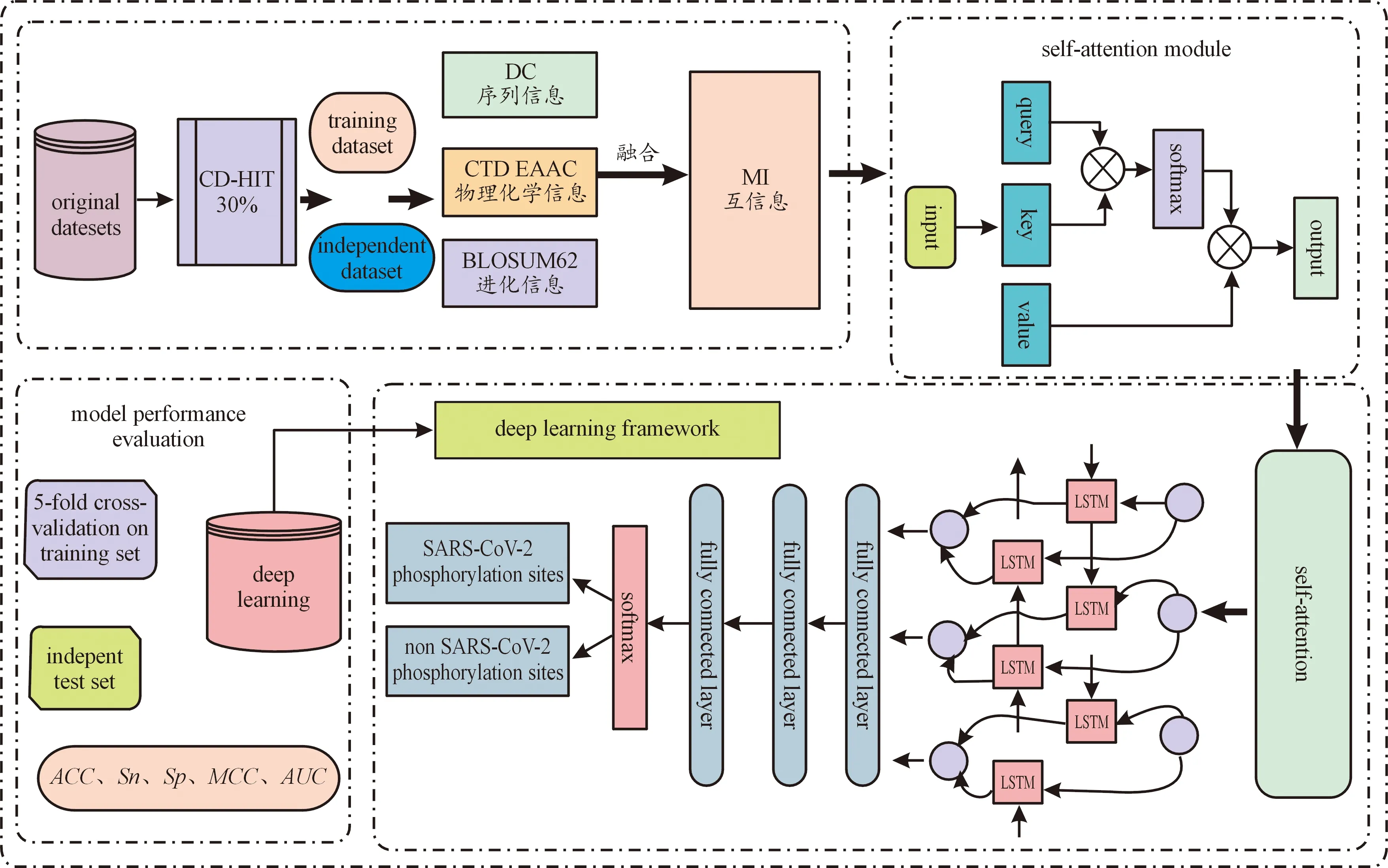
图1 Self-DeepIPs预测方法流程
Self-DeepIPs模型可以描述为以下几个步骤:
1) 获得训练集和测试集。数据包括新冠蛋白质序列及其对应标签,并严格按照按8∶2划分训练集和独立测试集。
2) 特征提取。利用4种特征编码方法 (DC、EAAC、CTD和BLOSUM62),将蛋白质序列信息转化为数字信息并进行多信息融合得到1 913维特征向量。
3) 特征选择。利用MI特征选择算法对融合的特征向量去除冗余和不相关信息。
4) 分类器。根据步骤2)、3),将所选取的最优特征子集以及所对应的类别标签,输入到SEBI分类器中预测新冠磷酸化位点。
5) 模型评估。根据步骤2)—4)中建立的模型采用五折交叉验证和独立测试集对模型进行评估。以ACC、Sn、Sp、MCC值作为评价指标,并绘制ROC曲线和PR曲线,并且使用独立的测试数据集对模型进行测试。
2 结果与讨论
2.1 特征提取和特征选择对结果的影响
在生物信息学中使用有效的特征方法对模型预测结果有重要的影响,然而使用单一的特征提取方法不能较好地说明新冠磷酸化位点的特征信息,采用4种特征提取方法(DC、EAAC、CTD和BLOSUM62) 进行多信息融合,并利用MI对多信息融合后的信息进行特征选择,然后将选择的最优特征子集输入到SEBI分类器中预测新冠磷酸化位点。各指标预测值的结果如表1所示。

表1 不同特征提取方法的预测结果比较
由表1可知,多信息融合4种特征提取方法后的ACC、Sn、Sp、MCC和AUC值分别为80.21%、80.13%、80.29%、60.42%和88.05%,均高于其他特征提取方法的指标。其中ACC值高出0.62%~6.12%,AUC值高出0.60%~6.23%。多信息融合之后再进行特征选择后的ACC、MCC和AUC值分别为83.62%、67.27%和91.70%。综合分析多个指标预测结果,多信息融合之后再进行特征选择能准确地提取蛋白质的信息,能更好地提高模型的预测性能。
2.2 分类器结果的影响
为了验证构建的分类模型(SEBI)的有效性,选用7种分类算法进行对比,其中包括AdaBoost[16]、NB[17]、RF[18]、XGBoost[19]、卷积神经网络(CNN)[20]、长短时记忆网络(LSTM)[14]和门控循环神经网络(GRU)[21],其中AdaBoost、NB、RF、XGBoost 4种分类算法均采用默认参数。CNN使用2个卷积层,一个最大池化层和一个完全连接的输出层。LSTM使用2个LSTM层和一个全连接的输出层。GRU使用2个GRU层和一个全连接层。SEBI使用一个自注意力层、2个BILSTM层和3个全连接层。8种分类模型的AUC和AUPR值如图2所示,SEBI与其他分类器的P值检验结果如表2所示。
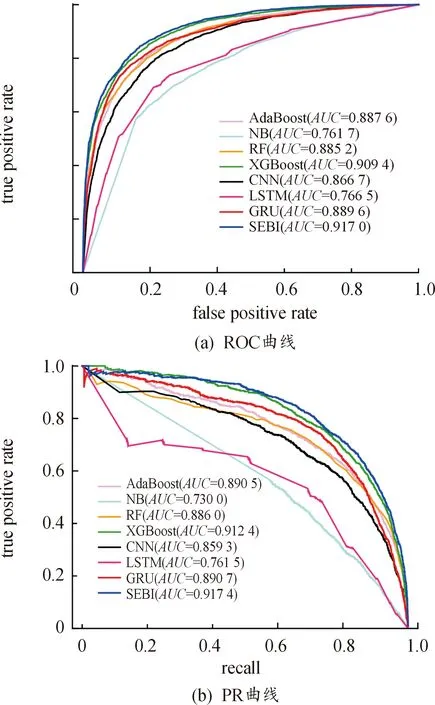
图2 对比不同分类器的ROC和PR曲线
由图2可以看出,SEBI分类器的ROC和PR曲线都明显覆盖了其他的分类器,AUC值和AUPR值分别达到91.70%和91.74%,其中AUC值高出其他分类器对应指标值的0.76%~15.53%,AUPR值高出0.50%~18.74%。因此,在S/T数据集上的ROC和PR曲线值均高于其他7个分类模型,达到最好的预测效果。因此构建的SEBI分类模型表现出更好的鲁棒性和泛化能力,能更准确地预测新冠磷酸化位点。
如表2所示,在显著性水平下,在ACC、MCC和AUC方面,构建的SEBI分类器在统计性检验上大多都优于AdaBoost、NB、RF、XGBoost、CNN、LSTM和GRU分类器,说明所构建的深度学习框架与其他分类器具备显著性差异,有较好的统计学意义。因此,采用SEBI分类模型作为识别新冠磷酸化位点的最佳分类器。
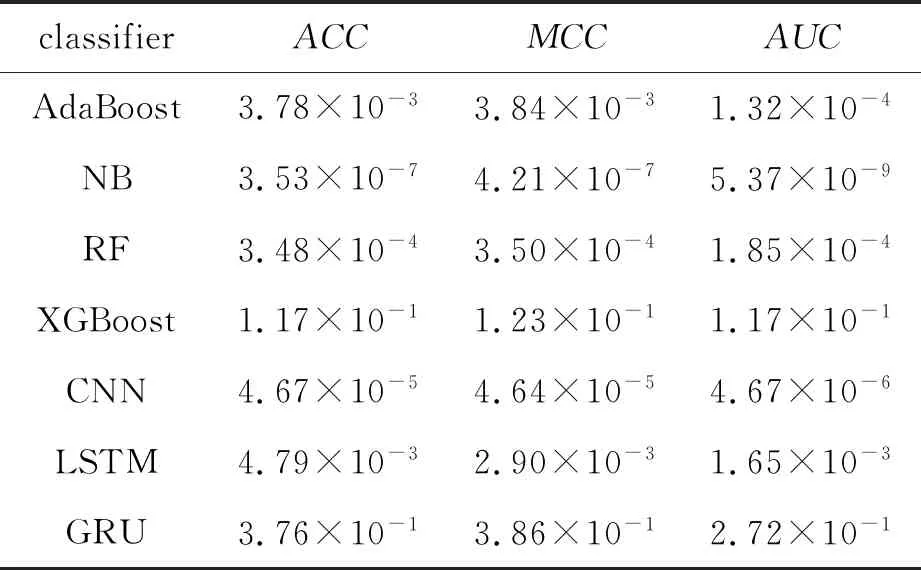
表2 SEBI分类器与其他分类器的P值 (T检验)
2.3 在独立测试集上本文方法与其他方法的比较
为了进一步评估Self-DeepIPs的性能,使用独立的测试数据将Self-DeepIPs与几种现有的磷酸化位点预测工具进行比较,这些模型分别为MusiteDeep2017[3]、DeepPSP[4]、DeepIPs[5]、MusiteDeep2020,如图3所示。
从图3中可以看出,构建的模型Self-DeepIPs在独立测试集上达到了较好效果,ACC和AUC值分别达到82.56%和91.23%,均超过了其他现存的磷酸化位点预测模型,不同指标结果表明提出的Self-DeepIPs预测模型明显提高了预测精度。说明构建的模型具有很好的鲁棒性和泛化能力,在很大程度上提高了磷酸化位点的预测性能。
3 结论
磷酸化位点的鉴定是了解SARS-CoV-2感染的分子机制和宿主细胞通路内变化的重要步骤。用于鉴定磷酸化位点的计算模型可以加速这些新型候选药物的开发。通过机器学习预测模型能极大减少实验鉴定新冠磷酸化位点的工作量,提出一种新的识别新冠磷酸化预测模型Self-DeepIPs,通过多信息融合蛋白质不同方面的信息,将蛋白质的序列信息转化为数字信息,然后利用MI去除冗余和不相关信息,首次使用自注意力机制和LSTM网络结合来构建深度学习框架识别新冠磷酸化位点。通过五折交叉验证和独立测试集对模型进行性能评估。在五折交叉验证下S/T训练集的AUC值达到91.70%,独立测试集上的AUC值达到91.23%。在训练集和独立测试集上的最终结果都达到了较好的预测效果。因此,提出的模型Self-DeepIPs能够有效预测新冠磷酸化位点,为实验鉴定新冠磷酸化位点提供更有意义的指导和帮助。
尽管构建的模型可以有效提高识别新冠磷酸化的预测精度,但仍有提升空间。下一步还要运用更新颖的深度学习方法对新冠磷酸化位点进行研究,扩大新冠磷酸化位点的数据集,更好地提高识别新冠磷酸化位点预测模型的精度,为实验鉴定新冠磷酸化位点的研究提供更有意义的指导。未来将尝试考虑蛋白质的结构信息,生成更有效的特征,同时考虑生物学意义,结合一些有效算法,比如图卷积神经网络,进一步提高Self-DeepIPs的预测性能。
猜你喜欢
天津医科大学学报(2019年6期)2019-08-13 07:04:42
电子制作(2018年19期)2018-11-14 02:37:08
电子测试(2018年1期)2018-04-18 11:52:35
自动化学报(2017年11期)2017-04-04 02:52:58
光学精密工程(2016年4期)2016-11-07 09:05:00
光学精密工程(2016年3期)2016-11-07 09:03:33
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
噪声与振动控制(2015年4期)2015-01-01 07:08:21
电测与仪表(2014年15期)2014-04-04 12:05:20
遗传(2014年3期)2014-02-28 20:59:01
