
土壤中9-芴酮和9,10-蒽醌的分析方法及赋存形态
2023-06-21 07:55古鹏汤佳豪赵庆庆王磊磊王加宁卢媛张闻
农业环境科学学报 2023年5期
古鹏,汤佳豪,赵庆庆,王磊磊,王加宁,卢媛,张闻*
(1.齐鲁工业大学(山东省科学院),山东省科学院生态研究所,山东省应用微生物重点实验室,济南 250103;2.南开大学环境科学与工程学院,环境污染过程与基准教育部重点实验室,天津 300350)
含氧多环芳烃(oxygenated polycyclic aromatic hy⁃drocarbon,OPAHs)是在化石燃料和生物质燃烧过程中或通过母体多环芳烃(PAHs)的光、化学和生物反应形成的一类新污染物,含有羰基、羟基或羧基等官能团[1-3]。OPAHs 可通过其极性官能团与土壤固相存在较强的相互作用,其稳定性高于其他含有硝基或烷基等基团取代的PAHs衍生物,因此OPAHs被称为“持久性终端代谢物”[4]。OPAHs 是可直接作用的诱变剂和致癌物,其生物毒性大于其母体PAHs,在不同环境基质均有检出,在部分地区其浓度高于衍生它们的亲代PAHs[1,4-5]。目前,人们在检测PAHs 类污染物时常集中于母体PAHs,对于和其同时产生或代谢产生的OPAHs关注度较低,且国内外对于OPAHs的研究主要集中于OPAHs的总量分析、源解析及毒性分析等[6],其赋存形态及生物有效性分析的相关报道较少。
OPAHs 在土壤中的赋存状态会影响其环境行为,进而影响其生态效应及环境风险,因此有必要对其赋存形态进行研究。连续提取法[7-11]被报道用于研究土壤中PAHs、脂肪烃等有机污染物的赋存形态,该法通过依次采用不同的提取步骤和试剂将与不同土壤组分结合的有机污染物区分为不同赋存形态,其中较详尽的分类包含生物有效态、难解吸态、富里酸结合态、胡敏酸态、粗胡敏素结合态、矿物结合态、干酪根结合态等7 种赋存形态,生物有效态与难解吸态的含量加和为有机溶剂提取态含量[10]。OPAHs 作为结构与PAHs 相似的有机污染物,其赋存形态分析亦适用于该法。连续提取法涉及从土壤相、水相和羟丙基-β-环糊精(hydroxypropyl-β-cyclodextrin,HPCD)相中提取目标物的过程[7-9],其中有机溶剂提取态、胡敏酸结合态、粗胡敏素结合态和干酪根结合态涉及从原始土壤或土壤组分(即土壤相)中提取的过程;富里酸结合态和矿物结合态涉及从不同试剂处理后含富里酸或矿物组分溶液(即水相)中提取的过程;生物有效态涉及从HPCD 溶液(即HPCD 相)中提取的过程。然而OPAHs 在以上各相中提取及测定方法尚不完备。Wietzoreck等[12]采用二氯甲烷-索式提取-气相色谱质谱联用法测定土壤中的OPAHs,Chibwe等[13]采用加压液体萃取-气相色谱质谱联用法测定土壤中的OPAHs,Hua 等[14]采用固相萃取-超高效液相色谱质谱联用法测定尿液中的羟基化PAHs。但索氏提取时间较长、有机溶剂消耗较大,加压液体萃取所采用的仪器昂贵,尚未普及,且测定采用气质或液质等色质联用仪器成本较高。因此,建立一种简便、高效、成本低廉的OPAHs提取及分析方法对研究OPAHs赋存形态具有重要意义。超声提取法和液液萃取法因其简便的操作和低廉的成本被广泛用于PAHs 在土壤相和水相中的提取,高效液相色谱(high performance liq⁃uid chromatography,HPLC)法是我国环保部发布的PAHs 的标准测定方法[《土壤和沉积物多环芳烃的测定/高效液相色谱法》(HJ 784—2016)]。于田田等[15]针对土壤/沉积物中的1-羟基芘建立了超声萃取-HPLC 测定方法。Martínez-Pérez-Cejuela 等[16]采用固相萃取-HPLC测定水相中硝化和氧化PAHs。曹笑语等[17]对OPAHs分析方法的研究进展进行了综述,认为气质或液质等色质联用法为目前主要的分析方法,关于HPLC分析方法的报道则比较有限,OPAHs仪器分析方法仍相对缺乏,更多种类的OPAHs在不同环境基质中简便且低成本的提取及测定方法有待建立和完善。
9-芴酮和9,10-蒽醌被多篇文献报道是土壤中丰 度 最 高 的OPAHs[1,18-20],本 研 究 以 这2 种 物 质 为OPAHs 代表物,建立其HPLC 分析方法及不同环境基质中的提取方法,利用连续提取法分析土壤中9-芴酮和9,10-蒽醌的生物有效态、难解吸态、富里酸结合态、胡敏酸态、粗胡敏素结合态、矿物结合态、干酪根结合态等7 种赋存形态的含量,探究土壤中不同OPAHs 形态赋存特征和自然衰减规律,旨在为控制土壤OPAHs污染提供理论依据。
1 材料与方法
1.1 供试材料
9-芴酮、9,10-蒽醌购自上海阿拉丁生化科技股份有限公司,纯度>98.0%,其结构式如图1 所示。二者分子量分别为180.21 和208.22,辛醇-水分配系数分别为3.58 和3.39,在25 ℃时的蒸气压分别为5.72×10-5Pa 和1.16×10-7Pa[21]。供试土壤采自山东济南(36°42'29″N,117°04'40″E),土地利用现状为耕地,土壤质地为壤土。其理化性质:pH 为8.25,有机质含量为5.2%,阳离子交换量为22.6 cmol·kg-1,总氮、总磷及总钾含量分别为2.28、0.96 g·kg-1和13 g·kg-1。土壤在室内自然阴干、磨碎过2 mm 筛后,向土壤中加入适量9-芴酮和9,10-蒽醌的混合丙酮储备液制备污染土壤。根据参考文献报道的实际污染土壤中[22]及相关方法学研究中[23]的OPAHs 含量,本实验9-芴酮和9,10-蒽醌在土壤中的含量都设置为20 mg·kg-1,即二者加和的OPAHs 污染水平为40 mg·kg-1。将污染土壤置于通风橱中2 d,每隔4 h 搅拌一次,使丙酮充分挥发后备用。
图1 9-芴酮及9,10-蒽醌分子结构式Figure 1 Molecular structure of 9-fluorenone and 9,10-anthraquinone
1.2 9-芴酮和9,10-蒽醌的HPLC分析方法构建
分别配制20 mg·L-1的9-芴酮和9,10-蒽醌丙酮溶液,采用二极管阵列检测器(diode array detector,DAD)进行全波长扫描(扫描范围190~400 nm)。全波长扫描使用的HPLC 条件为:流动相为甲醇∶水=9∶1,流速1 mL·min-1,进样量2 μL,色谱柱为Waters Symmetry C18(4.6 mm×250 mm,5 μm),柱温25 ℃。根据信号响应值,9-芴酮和9,10-蒽醌的最适检测波长分别为258 nm 和252 nm。在以上检测波长下,对HPLC 流动相配比及流速进行优化,筛选使得目标物的出峰时间合适,峰形尖锐、对称且分离度较好的流动相配比及流速。在此基础上建立OPAHs 测定外标曲线,配制1、2、5、10 mg·L-1和20 mg·L-1的9-芴酮和9,10-蒽醌系列混合标准溶液,进样分析,以峰面积为纵坐标,浓度为横坐标绘制标准曲线。
1.3 9-芴酮和9,10-蒽醌在不同基质中的提取方法构建
采用1.1 节制备的OPAHs 污染土进行超声提取实验,萃取剂选用二氯甲烷、甲醇和甲醇-二氯甲烷混合液(1∶1,V/V)。称取2 g 土,向其中加入10 mL 萃取剂,混合均匀后,超声10 min(功率500 W,频率40 kHz,25 ℃),3 000 r·min-1离心10 min,将上清液转移至50 mL 鸡心瓶中。上述操作重复3 次,合并萃取液至鸡心瓶中。将鸡心瓶旋蒸至近干,加入2 mL 甲醇定容,超声促溶后,溶液过0.45 μm 尼龙过滤器转移至自动进样小瓶中进行HPLC 检测,测定回收率,确定合适的萃取剂。
制备含有20 mg·L-1OPAHs 的水溶液及HPCD 溶液,其中9-芴酮和9,10-蒽醌质量比例为1∶1。萃取剂选用二氯甲烷和甲醇-二氯甲烷混合液(1∶1,V/V)。分别量取20 mL OPAHs 水溶液和HPCD 溶液于分液漏斗中,加入10 mL 萃取剂,进行液液萃取,然后将萃取液转移至50 mL 鸡心瓶中。上述操作重复3 次,合并萃取液至鸡心瓶中。其余操作同上。
1.4 土壤中9-芴酮和9,10-蒽醌赋存形态实验设计
分别称取10 g 1.1 节制备的OPAHs 污染土壤于多个40 mL 棕色玻璃瓶中,加入无菌水使其含水率为20.0%,用封瓶膜封口后置于生化培养箱中,25 ℃培养,每6 d 称量补水。在0、15 d 和30 d 进行有机溶剂提取态和生物有效态的提取与分析,并在30 d时进行全赋存形态的提取与分析。每个样品设置3个重复。
1.5 土壤中不同赋存形态9-芴酮和9,10-蒽醌的提取方法
土壤中OPAHs 赋存形态包括生物有效态(F1)、难解吸态(F2)、富里酸结合态(F3)、胡敏酸结合态(F4)、粗胡敏素结合态(F5)、矿物结合态(F6)和干酪根结合态(F7)。常报道的有机溶剂提取态(FDCM)包含F1和F2,本实验F1和FDCM分别单独提取,通过FDCM与F1 差减得到F2,F3~F7 在FDCM提取结束后的土样基础上连续分步提取获得。各态提取方法参照PAHs连续提取方法[7-8],并基于1.3 节9-芴酮和9,10-蒽醌在不同基质中提取方法的最优结果对其进行改进,具体如下:
生物有效态(F1):称取1 g冻干土样于玻璃瓶中,加入20 mL 用去离子水配制的50 mmol·L-1的HPCD水溶液,密封振荡(25 ℃,150 r·min-1)提取20 h,离心10 min(3 000 r·min-1,4 ℃)。收集上清液,用10 mL的二氯甲烷液液萃取3次,合并萃取液至鸡心瓶中。
有机溶剂提取态(FDCM):称取2 g 冻干土样于玻璃瓶中,加入10 mL 甲醇-二氯甲烷混合液(1∶1,V/V),混匀,超声10 min(功率500 W,频率40 kHz,25 ℃),离心10 min,将上清液转移至50 mL 鸡心瓶中。重复3次,合并萃取液至鸡心瓶中。
富里酸结合态(F3):将上一步有机溶剂提取结束后的土样37 ℃干燥2 h,添加10 mL 2 mol·L-1的NaOH 溶液,混匀,水解2 h(100 ℃)。水解结束冷却至室温,离心10 min,沉淀用于提取粗胡敏素结合态(F5),上清液用于提取富里酸(F3)和胡敏酸结合态(F4)。收集上清液,并用少量NaOH 溶液润洗土壤,离心后润洗液与上清液合并。用6 mol·L-1的盐酸溶液酸化上清液至pH<1,离心10 min,上清液为富里酸,沉淀为胡敏酸。将上清液转移至分液漏斗用10 mL 二氯甲烷液液萃取,重复3 次,合并萃取液至鸡心瓶中。
胡敏酸结合态(F4):向上一步得到的胡敏酸样品中加入无水硫酸钠除水,加10 mL 甲醇-二氯甲烷混合液(1∶1,V/V)超声提取10 min,重复3 次,合并萃取液至鸡心瓶中。
粗胡敏素结合态(F5):向富里酸结合态(F3)提取步骤中水解后的土样中加入10 mL 甲醇,振荡30 min,超声10 min,离心收集上清液;然后依次用10 mL甲醇-二氯甲烷混合液(1∶1,V/V)、二氯甲烷和二氯甲烷提取,步骤同上。将所有提取物合并,用20 mL 蒸馏水液液萃取,重复4 次,合并有机相至鸡心瓶中。提取后的土样用于矿物结合态(F6)的提取。
矿物结合态(F6):上一步得到的土样37 ℃干燥2 h,转移至50 mL 塑料离心管中,加入10 mL 6 mol·L-1盐酸溶液,振荡12 h,离心收集上清液;加入10 mL 40.0%氢氟酸-盐酸(1∶1,V/V),步骤同上。残渣用2 mol·L-1盐酸溶液润洗,离心后与上清液合并。收集的上清液用10 mL二氯甲烷液液萃取,重复3次,合并萃取液至鸡心瓶中。
干酪根结合态(F7):对矿物结合态(F6)提取后的土样,使用与提取粗胡敏素结合态(F5)相同的步骤进行处理,合并有机相至鸡心瓶中。
将鸡心瓶旋蒸至近干,加入2 mL 甲醇,超声促溶后,溶液过0.45 μm 尼龙过滤器转移至自动进样小瓶中待测。
1.6 9-芴酮和9,10-蒽醌分析
9-芴酮和9,10-蒽醌分析基于1.2 节的实验结果,具体为:通过HPLC 测定(安捷伦1260)。检测条件为:流动相为甲醇∶水=9∶1,流速1 mL·min-1,进样量2 μL,色谱柱为Waters Symmetry C18(4.6 mm×250 mm,5 μm),柱温25 ℃,DAD 检测器,检测波长252 nm(9,10-蒽醌)和258 nm(9-芴酮),检测时间6 min。
1.7 数据分析
通过SPSS 19.0 软件进行数据处理和单因素方差分析;使用Origin 2022制图。
2 结果与分析
2.1 9-芴酮和9,10-蒽醌的分析方法
通过反复试验流动相比例及流速,选择等度流动相甲醇∶水=9∶1,流速为1.0 mL·min-1,可在6 min内实现2种OPAHs完全分离。在此条件下,得到的色谱峰窄而尖,峰型均良好,无杂质峰干扰,如图2 所示。基于以上HPLC 测定条件,建立了9-芴酮和9,10-蒽醌的标准曲线,在1.00~20.00 mg·L-1范围内线性关系良好,相关系数R2达到0.999及以上,可满足测试需求。
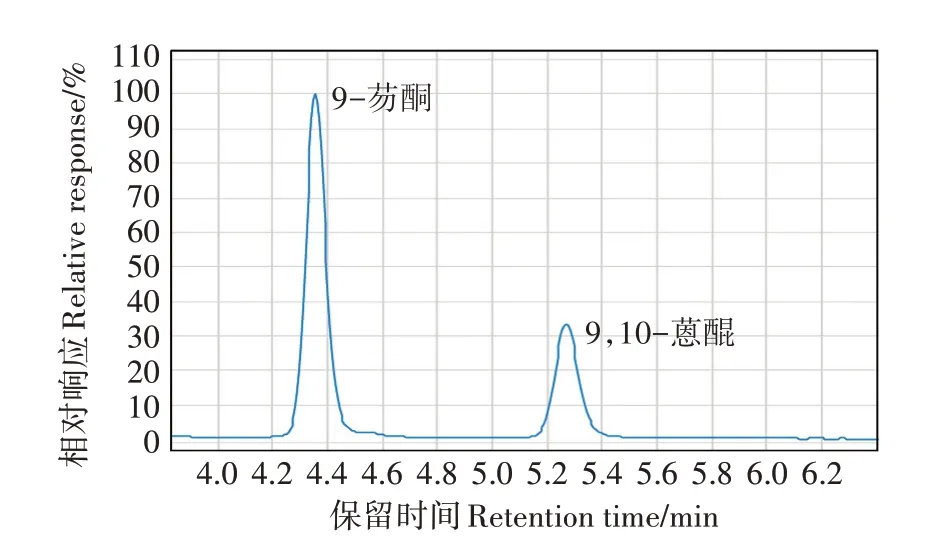
图2 9-芴酮和9,10-蒽醌的高效液相色谱图(检测波长258 nm)Figure 2 High performance liquid chromatography chromatogram of 9-fluorenone and 9,10-anthraquinone(detection wavelength 258 nm)
2.2 9-芴酮和9,10-蒽醌的提取方法
土壤相中的9-芴酮和9,10-蒽醌通过超声法进行提取,对比了3种萃取剂二氯甲烷、甲醇及甲醇-二氯甲烷混合溶液的萃取效率。土壤与萃取剂比例、超声时间及超声次数参考前期实验室从土壤中提取PAHs 的方法[24]。水相及HPCD 相中的9-芴酮和9,10-蒽醌通过液液萃取法进行提取,对比了2 种萃取剂二氯甲烷和甲醇-二氯甲烷混合溶液的萃取效率。液液萃取的比例、次数等参照从水相提取PAHs 的方法[25]。各基质中9-芴酮和9,10-蒽醌的提取回收率如图3 所示,对于土壤中这2 种OPAHs,使用混合萃取剂的提取效率最高,回收率为102.4%±0.8%(9-芴酮)和104.2%±3.4%(9,10-蒽醌),显著高于以二氯甲烷及甲醇为萃取剂的回收率。对于水相和HPCD 相中的这2种OPAHs,使用二氯甲烷作为萃取剂回收率最高,9-芴酮的回收率分别为87.8%±0.7%和86.3%±3.8%,9,10-蒽醌的回收率分别为87.2%±0.3%和78.7%±2.4%。
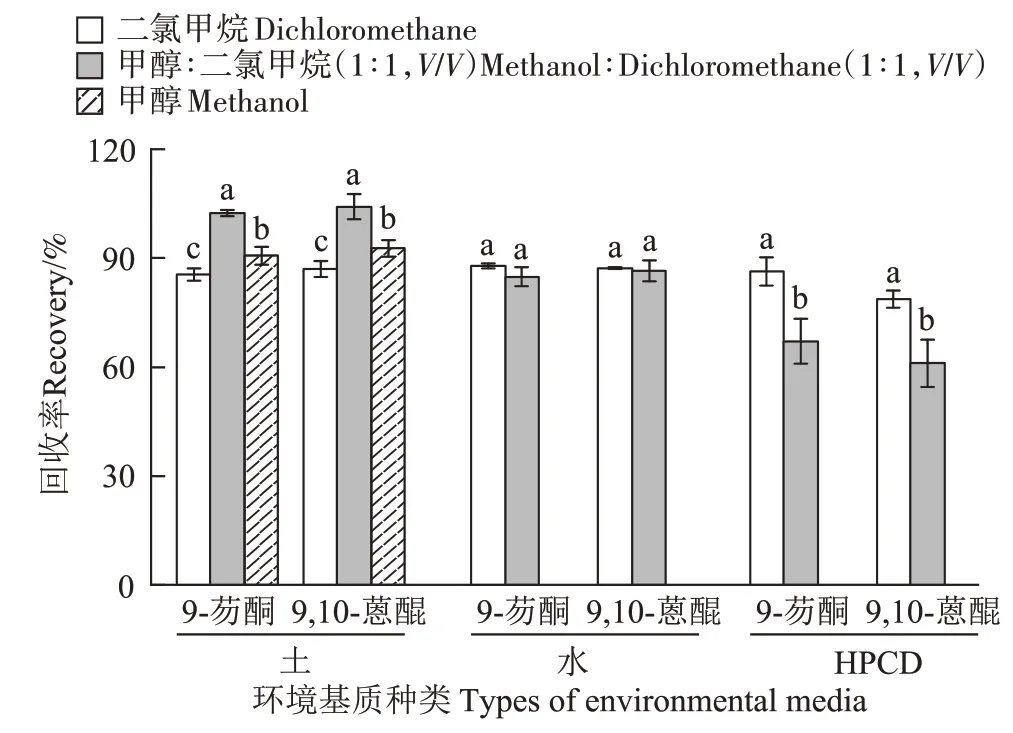
图3 土壤、水及HPCD相中的9-芴酮和9,10-蒽醌回收率Figure 3 Recovery of 9-fluorenone and 9,10-anthraquinone in the soil,water and HPCD
基于以上实验结果,最终确定土壤中9-芴酮和9,10-蒽醌最佳的提取条件为:以甲醇-二氯甲烷(1∶1,V/V)为萃取剂,土壤与萃取剂比例为2 g∶10 mL,超声时间10 min,超声次数3 次。水相及HPCD 相中9-芴酮和9,10-蒽醌的提取条件为:以二氯甲烷作为萃取剂,待提取液与萃取剂比例为20 mL∶10 mL,液液萃取3次。
2.3 9-芴酮和9,10-蒽醌在土壤中的赋存形态
图4为土壤被9-芴酮和9,10-蒽醌污染1个月后的各赋存形态含量,图5 为各赋存形态占总残留量的比例。二者反映了这2 种OPAHs 赋存形态的分布特征。同一种赋存形态下2 种OPAHs 污染物9-芴酮和9,10-蒽醌的含量之和用∑OPAHs表示。
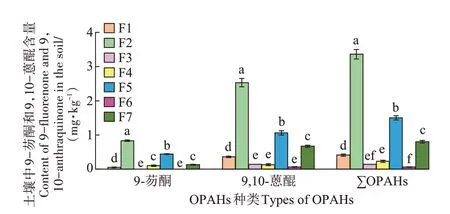
图4 土壤中9-芴酮和9,10-蒽醌及∑OPAHs各赋存形态含量Figure 4 Contents of 9-fluorenone,9,10-anthraquinone and∑OPAHs in soil
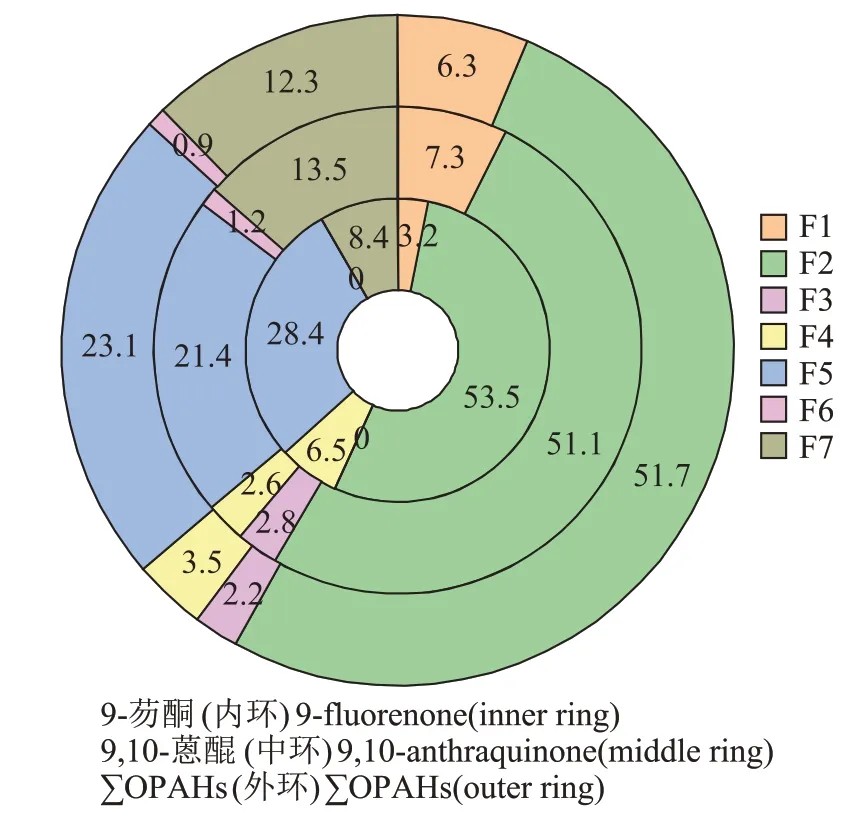
图5 土壤中9-芴酮和9,10-蒽醌及∑OPAHs各赋存形态含量占比Figure 5 Fractions of each speciation of 9-fluorenone,9,10-anthraquinone and ∑OPAHs in soil
9-芴酮的F1~F7 各态残留量分别为0.05、0.83、0、0.10、0.44、0 mg·kg-1和0.13 mg·kg-1,分别占其总残留量的3.2%、53.5%、0、6.5%、28.4%、0 和8.4%。9,10-蒽醌的F1~F7 各态残留量分别为0.36、2.53、0.14、0.13、1.06、0.06 mg·kg-1和0.67 mg·kg-1,分别占其总残留 量 的7.3%、51.1%、2.8%、2.6%、21.4%、1.2% 和13.5%。∑OPAHs 的F1~F7 各态残留量分别为0.41、3.36、0.14、0.23、1.50、0.06 mg·kg-1和0.80 mg·kg-1,分别占其总残留量的6.3%、51.7%、2.2%、3.5%、23.1%、0.9%和12.3%。综上可见,9-芴酮和9,10-蒽醌复合污染土壤中,9-芴酮各赋存形态残留量排序:F2>F5>F7>F4>F1>F3 和F6(F3 和F6 未检出);9,10-蒽醌排序:F2>F5>F7>F1>F3>F4>F6;∑OPAHs排序:F2>F5>F7>F1>F4>F3>F6。培养1 个月后,9-芴酮的各态残留量均低于9,10-蒽醌,9-芴酮、9,10-蒽醌、∑OPAHs的总残留量分别为1.55、4.95 mg·kg-1和6.50 mg·kg-1,分别占初始投加量的7.8%、24.8%和16.3%。
F1 及F2 态OPAHs 分别易于或较易于被生物利用,可直接或间接反映其在土壤中的有效性。二者之和为有机溶剂提取态,亦称为可提取态,9-芴酮和9,10-蒽醌可提取态含量之和为3.77 mg·kg-1,占这2 种OPAHs 在土壤中残留总量的58.0%,为土壤中存在的主要形态。F3 至F7 各态之和为结合态,是残留在土壤中不易被生物利用的部分,2种OPAHs结合态含量之和为2.73 mg·kg-1,占土壤中残留总量的42.0%。其中,F3、F4、F5 和F7 分别对应与土壤有机质组分富里酸、胡敏酸、胡敏素和干酪根紧密结合无法被有机溶剂直接萃取的OPAHs,这4 种形态OPAHs 之和为2.67 mg·kg-1,占结合态含量的97.8%。
2.4 9-芴酮和9,10-蒽醌生物有效性的变化
近些年来国内外十分关注土壤中有机污染物的生物有效性[26-27]。本研究中F1 直接反映9-芴酮和9,10-蒽醌在土壤中的生物有效性,FDCM是以往污染土壤调查中常用来表征污染物量的指标,因此对二者在30 d内的变化进行对比分析。
由图6 可知,9-芴酮和9,10-蒽醌的F1 和FDCM态在0 d 到30 d 的过程中均呈降低趋势,9-芴酮在0、15 d 和30 d 时F1 和FDCM态含量分别为9.56、0.08、0.05 mg·kg-1和14.85、1.86、0.88 mg·kg-1。9,10-蒽醌的相应含量分别为8.50、0.61、0.36 mg·kg-1和16.91、5.78、2.89 mg·kg-1。∑OPAHs相应含量分别为18.06、0.69、0.41 mg·kg-1和31.76、7.64、3.77 mg·kg-1。F1和FDCM在前15 d 降低极快,9-芴酮较0 d 分别降低99.2%和87.5%,9,10-蒽醌分别降低92.8%和65.8%,∑OPAHs分别降低96.2%和75.9%。F1 和FDCM在后15 d 降低趋势减缓,9-芴酮较0 d 分别降低0.3%和6.6%,9,10-蒽醌分别降低2.9%和17.1%,∑OPAHs 分别降低1.5%和12.2%。
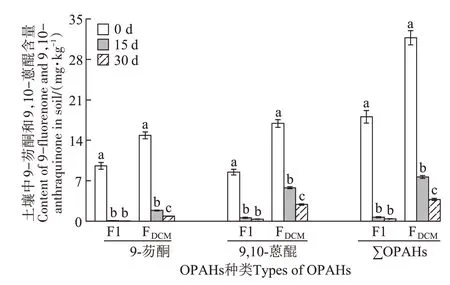
图6 不同时间点土壤中9-芴酮和9,10-蒽醌的F1和FDCM含量Figure 6 Contents of F1 and FDCM of 9-fluorenone and 9,10-anthraquinone in the soil at different time
3 讨论
3.1 不同基质中9-芴酮和9,10-蒽醌的萃取剂选择
OPAHs 萃取剂需与OPAHs 具有较大的亲和力,使其从土壤相或水相或HPCD 相进入萃取剂中,通过离心实现固液分离或静置实现液液分层。由于羰基的存在,9-芴酮和9,10-蒽醌的极性大于母体PAHs[28],提取时应选择具有一定极性且能形成氢键的溶剂。二氯甲烷作为萃取剂对PAHs 有较好的提取效果,但二氯甲烷为弱极性溶剂,无法与OPAHs 形成氢键,单独使用时回收率较低[15]。甲醇虽能与9-芴酮和9,10-蒽醌形成氢键,但是极性较强,对极性弱于甲醇的OPAHs 萃取率低;且甲醇黏度较大,扩散率较低,不易渗透至基质中与目标物结合,使目标物从基质中脱附,因而回收率较低。将甲醇与二氯甲烷混合之后,可以同时降低溶剂的极性和黏度,回收率明显提高。实验结果表明,对于土壤中的9-芴酮和9,10-蒽醌选用甲醇-二氯甲烷(1∶1,V/V)效果最好,回收率较二氯甲烷作为萃取剂分别高出17.0% 和17.3%,较甲醇作为萃取剂分别高出11.7%和11.5%。但是混合溶剂用于水相和HPCD 相中提取9-芴酮和9,10-蒽醌的提取效率低于二氯甲烷,可能是由于甲醇与水互溶,导致部分目标物残存于水相中,使其回收率较低。
3.2 9-芴酮和9,10-蒽醌在土壤中的赋存特征
土壤是高度不均匀的复杂体系,各组分间的疏水、静电相互作用、化学键的形成以及分子结构不同,使得污染物进入土壤后分化成不同的形态。污染物在土壤中的赋存形态直接影响其生态风险。目前,土壤中PAHs 赋存形态的研究较多,OPAHs 的赋存形态未见相关报道。聂婧等[29]研究了土壤中四环芳烃芘的赋存形态,各形态芘含量大小顺序为有机溶剂提取态>可脱附态>结合态,根据其提取方法,这三态分别对应本研究中的F2、F1、F3+F4,即F2>F1>F3+F4,F2和F1 是芘残留的主要存在形态,该规律与本研究9-芴酮和9,10-蒽醌的研究结果一致。Zhang 等[9]研究发现黄绵土中PAHs 的赋存形态为有机溶剂可提取态PAHs(59.1%)>胡敏素结合态PAHs(26.2%)>富里酸结合态PAHs(10.0%)>胡敏酸结合态PAHs(4.7%),对应本研究的赋存形态即为FDCM>F5>F3>F4。隋红等[30]研究发现PAHs在土壤有机质胡敏素组分中含量占比大,在富里酸和胡敏酸中占比相当少,即F5>F3 和F4。本研究中9-芴酮和9,10-蒽醌相应赋存形态含量顺序为FDCM>F5>F3或F4,与PAHs 研究结果一致。
本研究表明,9-芴酮和9,10-蒽醌进入土壤后会形成各种赋存形态,各赋存形态含量差异较大。∑OPAHs排序为F2(51.7%)>F5(23.1%)>F7(12.3%)>F1(6.3%)>F4(3.5%)>F3(2.2%)>F6(0.9%)。F1 与F2合并为可提取态,是9-芴酮和9,10-蒽醌在土壤中的主要赋存形态,可反映二者在土壤中的生物有效性,其中F2 是可提取态的主要组成部分。F3~F7 是9-芴酮和9,10-蒽醌与土壤有机质及矿物结合的部分。土壤有机质被报道是影响土壤中PAHs 赋存形态的重要因素[31-32]。本研究中土壤经有机溶剂提取后,残留的与有机质组分相关的OPAHs 赋存形态为F3、F4、F5 和F7,即富里酸、胡敏酸、胡敏素和干酪根结合态,其含量占比之和为41.1%;与矿物组分相关的赋存形态为F6,即矿物结合态,其含量占比为0.9%,有机质组分相关赋存形态含量远高于矿物结合态。因此,与PAHs 相似,2 种OPAHs 9-芴酮和9,10-蒽醌与土壤组分结合时,有机质组分较矿物组分发挥了更重要的作用。
土壤有机质根据其在不同pH值溶液中溶解度的差异,分为溶于任意pH值溶液的富里酸、溶于碱但不溶于酸的胡敏酸和既不溶于酸也不溶于碱的胡敏素[33]。富里酸和胡敏酸是溶解性有机质的主要组成部分[34],与其结合的9-芴酮和9,10-蒽醌即F3 和F4可迁移性较其他形态更强。2 种OPAHs 9-芴酮和9,10-蒽醌在富里酸和胡敏酸中含量分布表现不同,对于9-芴酮,F3
本文研究了9-芴酮和9,10-蒽醌2 种污染物在1种土壤中的赋存形态。OPAHs 除包含9-芴酮和9,10-蒽醌等含羰基的PAHs 衍生物以外,还包含PAHs分子中氢原子被1 个或多个羟基、羧基等官能团取代形成的羟基化及羧基化衍生物。这些OPAHs 既有芳香结构的相似性,又有含氧官能团的差异性,未来的研究应关注更多种类OPAHs 在更多不同性质土壤中的赋存形态,以评估其生态风险,支撑PAHs污染场地修复过程中OPAHs衍生物的监测、量化及风险管控。
3.3 9-芴酮和9,10-蒽醌生物有效性在自然衰减中的变化
土壤中的污染物通常可通过自然衰减实现土壤自净。Wei 等[41]研究表明,在未抑菌的芘污染土壤中,自然衰减初期,芘可提取率快速降低,后期则维持缓慢下降趋势。本研究发现,9-芴酮和9,10-蒽醌的F1 和FDCM含量在前15 d 降幅较大,后15 d 降幅趋缓,表明这2种OPAHs在自然衰减的不同进程,其生物有效性降低程度不同。Zhang等[11]的研究表明,PAHs及其硝基和羰基衍生物进入土壤30 d 后的可提取率及生物有效性明显下降。9-芴酮和9,10-蒽醌生物有效性降低的主要原因可能是:一方面,自然挥发和微生物降解等作用可导致土壤中这2 种OPAHs 含量降低;另一方面,随着9-芴酮和9,10-蒽醌与土壤接触时间的增加,这2种OPAHs逐渐老化进入土壤有机质和矿物质等组分中形成结合态残留。
0 d 时,土壤中9-芴酮的F1 态含量高于9,10-蒽醌,15 d 和30 d 时反之,且在30 d 时9-芴酮的各赋存形态含量均低于9,10-蒽醌,即9-芴酮较9,10-蒽醌在土壤中自然衰减得更快,这与两者的性质密切相关。9-芴酮的蒸气压高于9,10-蒽醌,更倾向于从土壤相向气相中逸散;此外,9-芴酮的水溶解度高于9,10-蒽醌,更易被土壤微生物利用,使得土壤中9-芴酮的残留量更低。Idowu 等[42]研究了极性PAHs 的环境健康风险,指出高分子量化合物比低分子量化合物通常表现出更大的毒性,且具有氧和氮杂原子的化合物由于更高的反应性,其毒性比同环烃更大。Trine等[43]研究发现杂酚油污染土壤蒸汽强化萃取修复过程中形成了PAHs衍生物,分子量较低的OPAHs 1,4-萘醌会比分子量较高的OPAHs 9,10-蒽醌降解得更快,即其生物有效性更强。本研究中9-芴酮的分子量小于9,10-蒽醌,推测9-芴酮具有更快的降解速率。Qiao 等[44]总结了OPAHs 的生物降解半衰期,9-芴酮和9,10-蒽醌的生物降解半衰期分别为8.98 d和16.7 d,9-芴酮的半衰期小于9,10-蒽醌,说明9-芴酮更易被生物降解。
4 结论
本文针对2 种含氧多环芳烃(OPAHs)9-芴酮和9,10-蒽醌,建立了HPLC-DAD 测定方法及其在不同基质中的提取方法。土壤中这2 种OPAHs 提取使用超声提取法,萃取剂选用甲醇-二氯甲烷(1∶1,V/V)混合液;水和HPCD 溶液中这2 种OPAHs 提取使用液液萃取法,萃取剂选用二氯甲烷。
进入土壤30 d后,各赋存形态9-芴酮和9,10-蒽醌含量之和大小顺序为难解吸态>粗胡敏素结合态>干酪根结合态>生物有效态>胡敏酸结合态>富里酸结合态>矿物结合态,可提取态和结合态占总残留量的比例大致相当,有机质结合态较矿物结合态含量占比更高。随着自然衰减进行,9-芴酮和9,10-蒽醌的生物有效性降低,9-芴酮较9,10-蒽醌在土壤中生物有效性降低得更快。
猜你喜欢
湖南农业科学(2022年11期)2022-12-20
原子能科学技术(2021年6期)2021-06-30
中成药(2018年10期)2018-10-26
天然产物研究与开发(2018年8期)2018-09-10
中成药(2018年4期)2018-04-26
广东教育·高中(2017年11期)2017-12-04
腐植酸(2015年1期)2015-04-17
中国当代医药(2015年23期)2015-03-01
中国果业信息(2015年1期)2015-01-24
植物营养与肥料学报(2012年5期)2012-10-26
