
西藏山溪鲵转录组组装与分析
2023-06-20 04:51:58朱文文熊建利
渔业研究 2023年3期
朱文文,郁 川,熊建利,黄 勇
(1.洛阳职业技术学院食品与药品学院,河南 洛阳 471000;2.河南省动物疾病与公共卫生工程研究中心,河南 洛阳 471000;3.绵阳师范学院,生态安全与保护四川省重点实验室,四川 绵阳 621000;4.河南科技大学动物科学院水生动物适应与进化实验室,河南 洛阳 471000)
西藏山溪鲵(Batrachuperustibetanus)隶属有尾目(Caudata)、小鲵科(Hynobius)、山溪鲵属(Batrachuperus),主要分布在青海、甘肃、陕西、四川、西藏等海拔1 500~4 000 m、植被较为丰富的山区溪流中或泉水石堆下[1-2]。西藏山溪鲵是我国珍贵、稀有的濒危水生野生动物,已被列入《国家保护的有益的或者有重要经济、科学研究价值的陆生野生动物名录》。由于西藏山溪鲵具有很高的药用价值,其也被列入我国传统藏药药典;同时具有重要的科研和生态价值[3]。近年来,由于环境恶化、生态条件退化和人为过度开发利用该物种资源,导致其生存空间正在逐渐缩小,种群数量明显减少,在IUCN(International Union for Conservation of Nature)红色名录中被列为易危的物种[4]。目前,有关西藏山溪鲵转录组的研究尚未见报道。而了解西藏山溪鲵转录组基因信息,能为后续科学合理地利用与保护该物种基因资源提供理论基础。
转录组高通量测序是近几年发展起来的新技术,即RNA-Sequencing(RNA-Seq),具有处理数据量大、运行成本低、灵敏度高等优点,现已成为发掘物种功能基因的重要研究手段之一[5-6]。该高通量技术可在没有该物种已知全部基因组序列信息的前提下,在较短时间内准确地得到特定组织或者细胞在特殊状态下全部的转录组信息,能完整识别该条件下已知基因的表达、生理状态与特定的分子机制调控过程,并能辨别一些未知的转录本和遗传标记信息等,在非模式生物转录组的研究中得到了广泛应用,为进一步研究生物学提供了更全面、便利的平台[7-9]。目前,有关山溪鲵属物种的研究均集中在属种形态分类、系统发育与进化和生理生化等方面[10-15]。为挖掘西藏山溪鲵基因数据和功能,本研究利用高通量测序技术对西藏山溪鲵进行转录组测序,并结合现代生物信息学分析方法对测序得到的序列进行拼接、组装和功能注释分析。得到的数据有利于更全面了解西藏山溪鲵转录组信息,方便科技工作者实现数据资源共享,为后期开展西藏山溪鲵分子遗传学及生物多样性研究提供基础资料。
1 材料与方法
1.1 材料
2017年7月,从四川百灵山采集4只外表无伤、体长130~150 mm的西藏山溪鲵成体(2♂、2♀)为研究对象。
1.2 样品收集
利用MS-222将标本麻醉后,分别取4尾成鲵的脾脏、肌肉、肾脏、肝脏、心脏、肠道、皮肤和性腺组织,每个组织取样约10 mg,最后将所有组织样品混为1个样品,于-80℃超低温冰箱保存、备用。
1.3 组织总RNA检测
将混合的西藏山溪鲵组织样品迅速置于装有液氮的研钵中,研磨成粉状,根据Takara公司提供的Trizol Reagent操作说明书完成总RNA的抽提。获得的总RNA 再经过1.2%琼脂糖凝胶电泳和Nanodrop-2000核酸蛋白测定仪检测总RNA的完整性和纯度。经检验合格的总RNA样品再进行后续的转录组测序。检测标准定义为:总RNA量≥10 μg,OD260/280为1.75~2.10,28 S∶18 S≥1.5∶1.0,RIN值≥8.0。
1.4 转录组测序与分析
样品检测合格后,利用 Oligo (dT)磁珠法纯化出mRNA,然后将mRNA截为片段,经过PCR方法得到西藏山溪鲵cDNA文库,最后建好的cDNA文库利用Illumina HiSeq 4000平台技术进行上机测序,由杭州联川生物技术股份有限公司完成测序。原始 reads 经过去接头并且过滤掉低质量及长度过短序列后,得到高质量测序数据(clean reads)。利用 Trinity 软件的Paired-end拼接方法对clean reads进行Denovo组装,得到unigenes。由于西藏山溪鲵目前没有基因组数据,本研究以报道的墨西哥蝾螈(Ambystomamexicanum)(https://www.ncbi.nlm.nih.gov/data-hub/genome/GCA_002915635.3/)基因组序列作参考基因组,使用STAR 软件进行比对[16],再用StringTie 软件基于参考基因组注释文件对所有转录本进行整合组装[17]。使用MiscroSAtellite(MISA)软件进行SSR鉴定和分析。应用Rsem软件进行基因表达定量分析,基因表达量采用FPKM值(Fragments per kilobase of transcript per million fragments mapped)表示。最后,利用Swiss-prot (Swiss prot protein database)、Nr (Non-redundant protein sequences)、KEGG (Kyoto encyclopedia of genes and genomes)、KOG (Eukaryotic ortholog groups)和 Pfam (Protein families database)和GO (Gene ontology)6个公共数据库与组装得到的西藏山溪鲵unigenes进行Blast序列比对[18]。选择阀值条件为E value <1e-10,进行功能注释。
2 结果与分析
2.1 RNA检测
提取的混合组织总RNA 样品呈现完整清晰的28 S、18 S和5 S带型,OD260/280值为2.03,28 S∶18 S值接近1.80,且RIN值为8.0,说明提取到的总RNA质量较高,符合后续转录组测序建库要求。
2.2 测序质量控制
采用Illumina Hiseq 4000测序得到西藏山溪鲵转录组的数据。对获得的原始数据经过测序质量控制,得到49 924 038 bp的clean reads,包含7.49 G的碱基数据。测序质量显示:碱基Q20(序列质量不低于20的碱基所占百分比)占96.99%,Q30占93.61%(>85%),GC含量平均值为47.18%,表明测序碱基组成的结果较好,组装质量完整性较高,能用于下一步分析。
2.3 转录组数据组装与分析
在去除低质量数据和进行质控后得到的clean reads,采用Denovo方法进行序列拼接后,总共获得43 626条转录本,序列长度为46 293 115 bp,其中N50片段序列的长度为1 822 bp,平均长度为1 061 bp。在获得转录本序列的基础上,经Trinity软件进行组装,参数选用 Trintity 的省缺参数 Kmer=25[19],然后拼接好的片段进一步合成。最终得到了36 252条unigenes,共33 976 485 bp,序列大小范围为201~20 766 bp,得到的平均长度为937 bp,其中N50为1 549 bp(表1)。对每条unigenes长度统计相应的unigenes数量,其中长度范围在200~500 bp的最多,有16 775条,占总数的46.27%;其次长度在500~1 000 bp之间的有9 091条,占总数的25.1%;长度在1 000~2 000 bp之间的有6 263条,占总数的17.28%;长度大于2 000 bp以上的最少,有4 123条,占总数11.37%,随长度增加基因数量逐渐减少,说明测序的序列、数据拼接和组装质量较高。
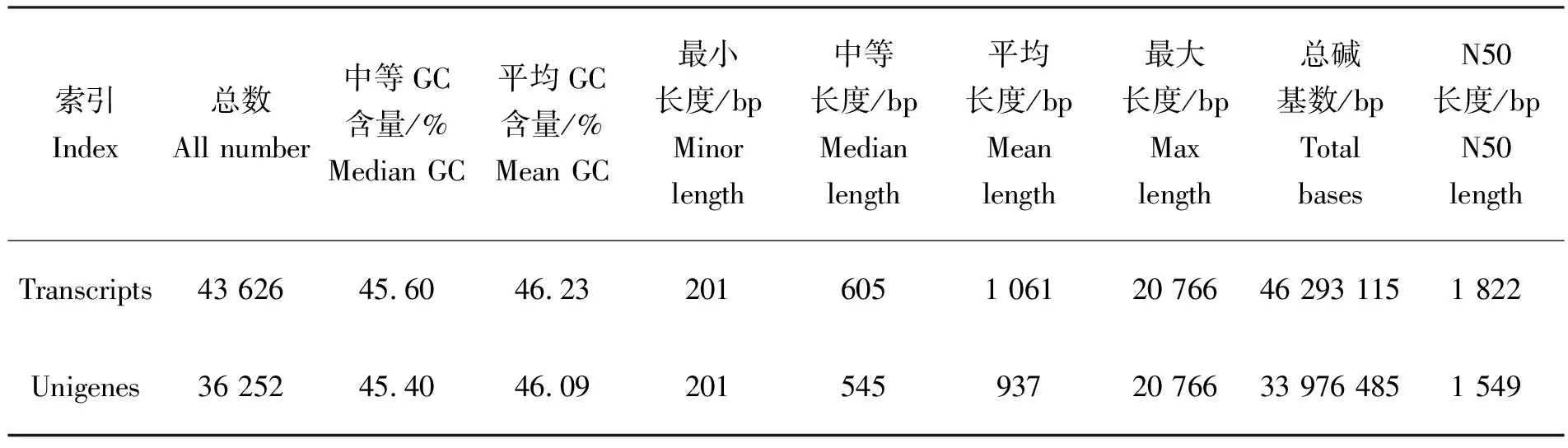
表1 测序组装的序列结果统计
2.4 SNP检测分析
单核苷酸多态性定义为基因组上单个核苷酸产生的变异,是进行分子标记鉴定、辅助育种和遗传图谱构建等非常重要的一种遗传标记方法。本研究利用转录组作为参考序列,使用BWA和Samtools软件对西藏山溪鲵的外显子区域进行SNP发掘,结果显示总共得到 3 100个SNP 位点,包括A-G、C-T、A-C、A-T、C-G和G-T六种颠换类型的SNP。在所有颠换类型的SNP中,A-G和C-T两种颠换类型的比例最高,占所有SNP位点的58.94%;C-G颠换类型占的比例最低,仅为6.87%。A-C、A-T和G-T这3种 SNP颠换类型有相似的比例,占总量的34.19%(表2)。
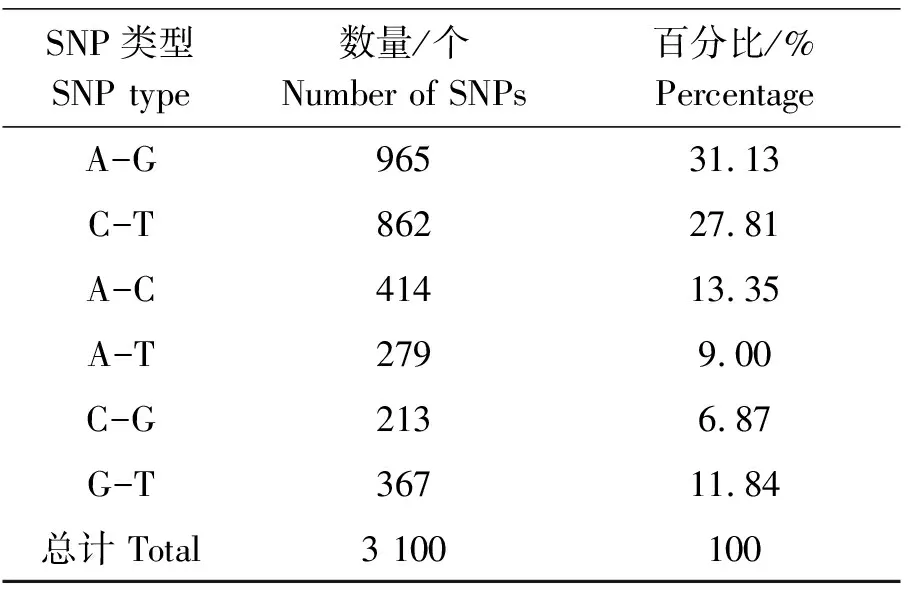
表2 SNP位点的分类
2.5 CDS预测与微卫星SSR位点分析
通过Genscan软件预测其CDS序列,预测结果显示:总共检测出17 472条unigenes可被编码,占全部36 252条unigenes的48.2%,未检测到CDS的unigenes有18 780条。长度在200~500 bp所编码的氨基酸占比最高,预测到超过2 000 bp的编码氨基酸序列有1 045条,能编码氨基酸平均长度为367.6 bp。使用MISA软件对36 252条unigenes进行搜索SSR位点,总共有2 779条序列被检测到具有SSR位点(表3)。其中,单核苷酸至六核苷酸重复类型均有被检测到,SSR类型单核苷酸出现频率最多,有1 473个;其次为二核苷酸,有715个;490个SSR具有三核苷酸位点;四和五核苷酸SSR位点的数量分别为56个和38个;SSR位点为六核苷酸的数量最少,仅为7个。这些SSR位点可为后续进行分子标记鉴定提供引物设计基础。
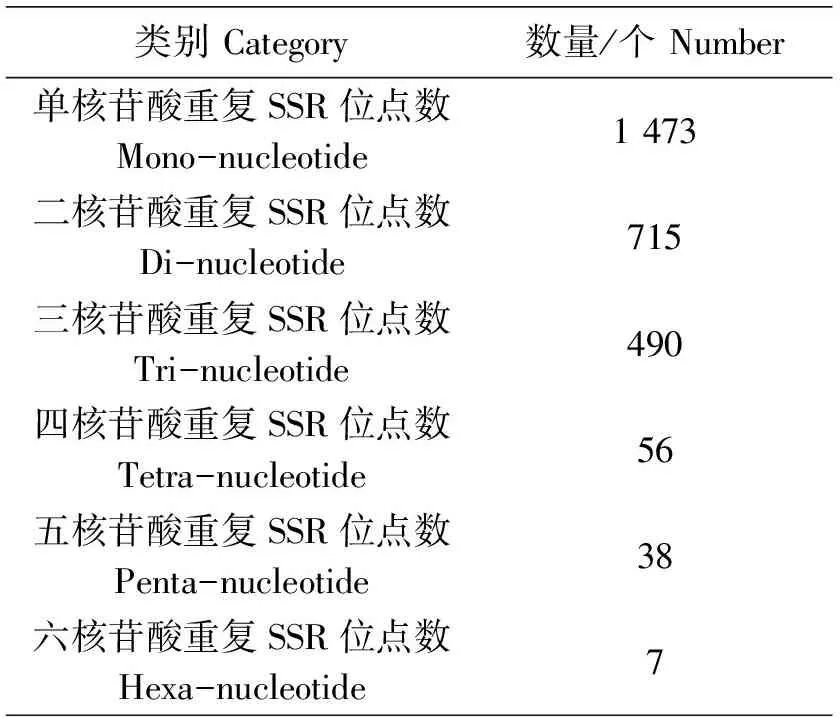
表3 SSR位点统计
2.6 基因表达量分析
在转录组测序中,用RPKM方法计算基因的表达水平(表4),表示在每百万reads中来自某一基因每千碱基长度的read读数。本研究以RPKM≥0.1作为基因表达标准,在已获得的36 252个unigenes中,有基因表达量的序列为36 226条。对不同RPKM 区间的基因数量进行统计发现,其中RPKM值在3.57~15.00的基因最多,为17 099条,占到了47.2%;其次是RPKM值位于0.30~3.57的基因,数量为13 868条;RPKM值位于15~60的基因有4 000条,高表达的基因RPKM值>60为1 243条;而RPKM值在0.1~0.3的低表达基因最少,仅16条,占所有表达基因的0.04%。上述结果表明Illumina HiSeq测序技术能够检测到极低水平基因的表达。
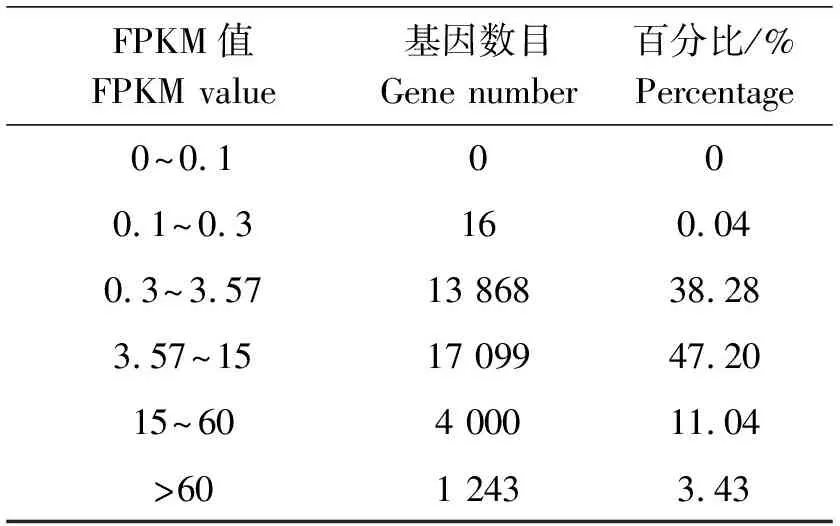
表4 FPKM值密度分布
2.7 Unigenes功能注释
在6个数据库中对组装得到的36 252条unigenes进行Blast比对,并进行功能注释。注释成功的unigene基因数目在不同数据库中所占比例有所差别。如表5所示,在Swiss-port数据库获得注释的unigenes有16 465条,占总数的45.42%;在Nr数据库中获得注释的unigenes有18 749条,注释比例最大,达到了51.72%;在Pfam数据库中获得注释的unigenes有13 983条,占38.57%;在KEGG数据库注释的unigenes有10 607条,占29.26%;在KOG数据库中获得注释的unigenes有15 704条,占43.32%;在GO数据库中获得注释的unigenes有14 242条,占39.29%。在Nr数据库中,按物种分布统计,与西部锦龟(Chrysemyspicta)匹配度最多,为13.6%;其次是热带爪蟾(Xenopustropicalis)和绿海龟(Cheloniamydas),分别为9.5%和7.8%,最低的是非洲爪蟾(Xenopuslaevis),为4.2%,而与其他物种蛋白质无匹配的unigenes占55.8%(图1)。
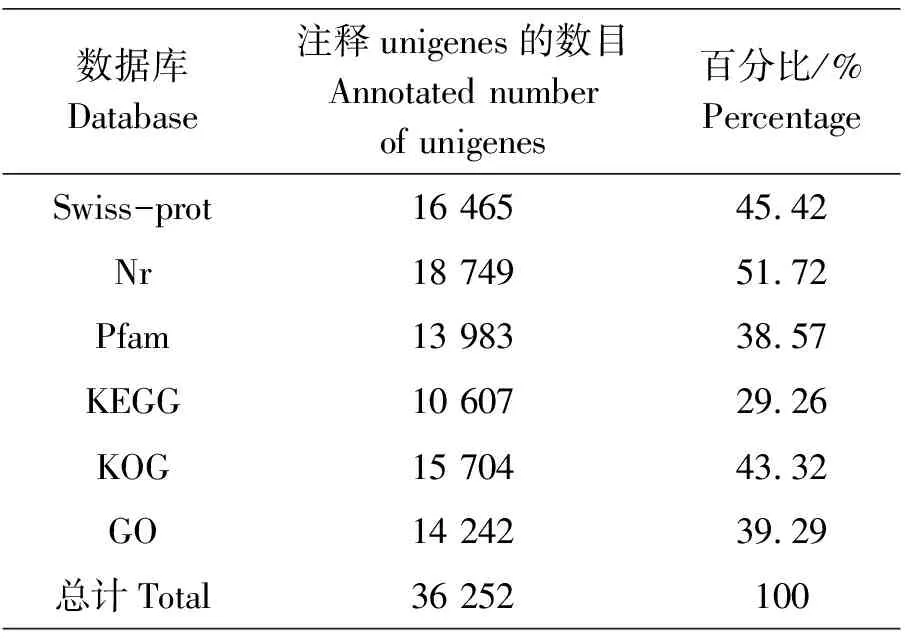
表5 序列功能注释

2.8 KOG功能分类
将获得的西藏山溪鲵unigenes 在COG数据库中进行功能注释,可分为25类(图2),在COG中注释到的unigenes涉及功能类别较广,与生命活动相关的占大部分。其中,基因数注释最多的是一般功能预测类,有2 490条。其次,信号转导机制与翻译后修饰、蛋白质周转、伴侣类基因,分别有2 210条和1 140条;表明遗传信息的传递在西藏山溪鲵生理活动中极为活跃。值得注意的是,注释到细胞运动类基因最少,仅有40条,这说明该物种与它们高海拔独特的生活环境和迁徙能力弱有关。
2.9 GO功能注释
根据得到的注释信息进行分类,共有14 242条unigenes被注释,占39.29%。按照GO功能分类方式将注释到的unigenes主要分为3大类(生物过程、细胞组分和分子功能),如图3所示。这3个大类别又被详细地划分为50个功能亚类小组。其中大类生物过程包含25个不同的亚类功能组,这也是三大类中所含功能类别最多的一类,注释到转录、DNA依赖性和转录调控、DNA依赖性的unigenes占最多,分别有1 183条和932条,占比分别为72.27%和60.13%;而与RNA剪接相关unigenes所占数量最少,仅140条,占12.02%;在细胞组分中,有15个亚类,注释到细胞核的unigenes最多(2 798条),占90.12%;注释到细胞膜的unigenes最少,仅为138条。分子功能类别中又划分为10个亚类,注释到ATP结合相关功能的unigenes数量最多,有1 855条,其次是注释到锌离子结合的unigenes,有1 668条。而注释到蛋白丝氨酸/苏氨酸激酶活性相关的unigenes数量最少,有377条序列。
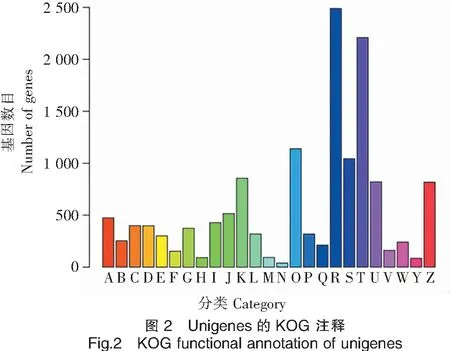
注:A.RNA 加工和修饰;B.染色质结构与动力学;C.能量产生和转换;D.细胞周期调控、细胞分裂、染色体;E.氨基酸运输和代谢;F.核苷酸运输和代谢;G.碳水化合物的运输和代谢;H.辅酶运输和代谢;I.脂质运输和代谢;J.翻译、核糖体结构和生物合成;K.转录;L.复制、重建和修复;M.细胞壁/细胞膜/膜结构的生物合成;N.细胞运动;O.翻译后修饰、蛋白质周转、伴侣;P.无机离子转运与代谢;Q.次生代谢产物的合成、转运和代谢;R.普通功能预测;S.未知功能;T.信号转导机制;U.胞内运输、分泌和囊泡运输;V.防御机制;W.胞外结构;Y.核结构;Z.细胞骨架。
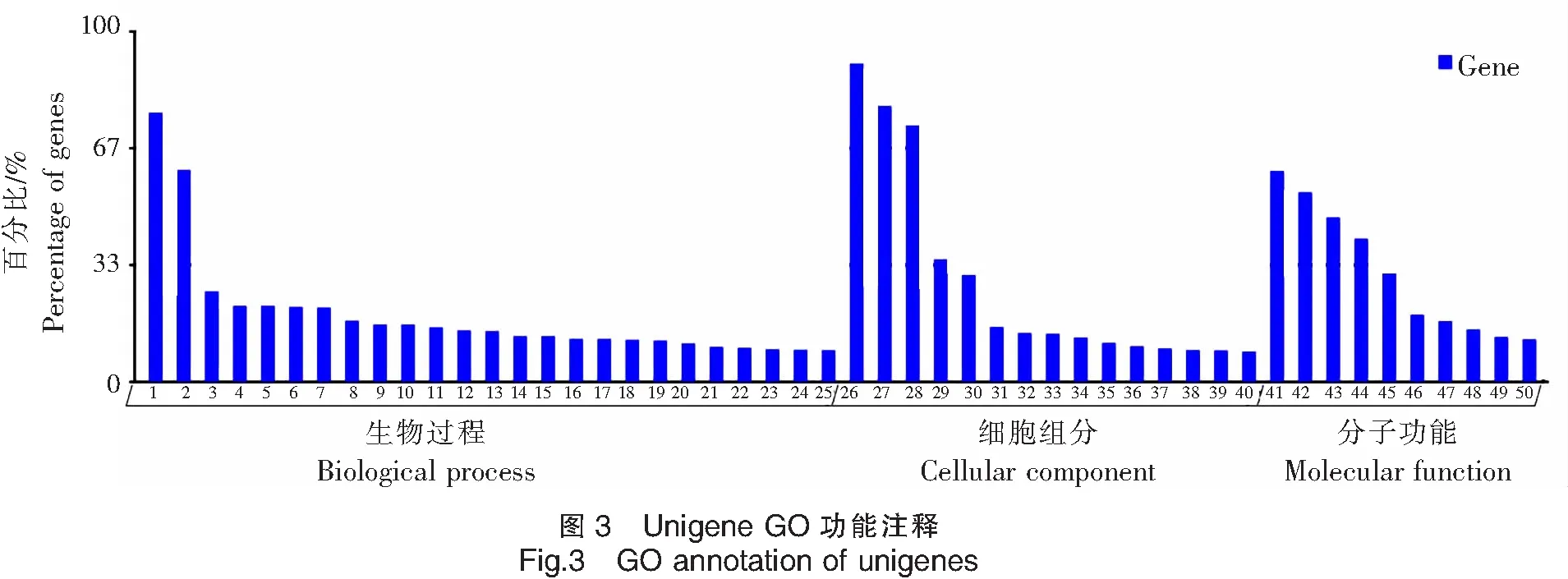
注:1.转录、DNA依赖性;2.转录调控、DNA依赖性;3.蛋白质转运;4.多细胞器官发育;5.细胞分化;6.凋亡;7.细胞黏附;8.蛋白质水解;9.信号转导;10.细胞分化;11.细胞周期;12.有丝分裂;13.小G蛋白介导信号转导;14.DNA 修复;15.转运; 16.RNA加工;17.翻译;18.转录正调控;19.胞内信号转导;20.染色质修饰;21.转录负调控;22.细胞内蛋白质转运;23.跨膜输送;24.精子形成;25.RNA剪切;26.细胞核;27.必需膜;28.细胞浆;29.胞液;30.细胞质膜;31.内质网膜;32.线粒体;33 胞外区;34.胞核;35.核质;36.细胞骨架;37.高尔基体膜;38.微管;39.高尔基氏复合体;40.细胞膜;41.ATP结合;42.锌离子结合;43.蛋白质结合;44.DNA 结合;45.金属离子结合;46.RNA结合;47.钙离子结合;48.结合;49.特异序列DNA结合;50.蛋白丝氨酸/苏氨酸激酶活性。
2.10 KEGG功能分类
对西藏山溪鲵所有的 unigenes基因进行KEGG通路注释,共有10 607条 unigenes 得到注释,这些注释到的unigenes涉及到生物系统、代谢、遗传信息处理、环境信息处理和细胞过程5个大类30亚类的通路信息。如图4 所示,这些注释到unigenes 分布于257个已知功能的代谢通路中,其中有较多的unigenes涉及信号转导通路,共有1 058条;其他的几个分别为MAPK 信号通路(320条)、Wnt信号通路(191条)、Calcium信号通路(190条)、ErbB信号通路(130条)和TGF-beta信号通路(118条),这些代谢通路都与环境信息处理大类中信号转导相关。注释到细胞通讯通路的unigenes有862条与细胞过程有关,占第二位;其次是注释到与免疫系统通路相关的unigenes有757条;推测这与西藏山溪鲵在遭受到病原微生物入侵中产生的免疫应答过程有着重要功能,同时也富集到与环境适应相关的unigenes,为28条,这可能与西藏山溪鲵适应高山生活环境的特殊性有一定关系。
3 讨论
目前高通量测序技术在缺少基因组信息的非模式生物研究中已被广泛应用[20-23]。当前尚无西藏山溪鲵转录组研究工作的报道。本研究中,利用这种测序技术对西藏山溪鲵组织进行了转录组测序,在没有参考基因组的情况下对其进行了拼装,共产生了36 252条unigenes,检测到的unigenes平均长度为937 bp,其中长度≥2 000 bp以上的有4 123条。表明本次测序质量较高,数据组装效果很好;也说明高通量测序是一种可靠性较好、能高效获取非模式生物基因序列的方法。对所有获得的unigenes在6大数据库中进行比对,结果都得到了注释。但在物种注释上,注释结果相似度最高的是西部锦龟,也仅为13.6%;仍有10 562条(55.8%)unigenes注释的物种不明确,一方面原因可能是基因数据库中山溪鲵属物种基因资源偏少,影响功能注释的基因;另一方面可能有些unigenes是西藏山溪鲵特有的基因,后续需要进一步的研究。
在GO注释的unigenes中,与转录、DNA依赖性、细胞核和ATP结合相关的基因最多,这可能与西藏山溪鲵组织的生长、细胞的增殖与分化和能量代谢密切相关。在KOG注释的unigenes中,获得了西藏山溪鲵转录组数据库,得到了与西藏山溪鲵生长发育、生物合成与代谢相关基因资源。此外,从KEGG通路分类结果看:共有10 607条西藏山溪鲵unigenes参与到257个已知功能的代谢通路中。其中参与西藏山溪鲵信号通路的unigenes最多,为1 058条,其中最多的是MAPK 信号通路和Wnt信号通路的unigenes数目很多,分别为320条和191条,说明这些信号分子在西藏山溪鲵生命活动与代谢活动中起着重要的生理作用。其次是参与细胞通讯通路的unigenes有862条,这可能与西藏山溪鲵在生长过程中要不断适应自身环境有关。其中unigenes被注释到免疫系统通路中占第三,有757条。这些涉及免疫通路相关的基因主要包括Toll样受体、T细胞抗原活化分子、干扰素刺激基因模式识别受体、补体成分和抗菌肽基因等,表明在西藏山溪鲵生长中可能形成了其特有的天然免疫机制。本研究还发现有48条unigenes注释到环境适应,推测可能与西藏山溪鲵特定的低温栖息环境和适应高海拔生活特点有关。
此外,高通量测序技术的另一个优势是能快速地从大量基因序列中获得SSR分子标记资源,能被广泛用于动植物的进化论和遗传学研究[20]。本研究中利用MISA软件查找测序的数据,检测到SSR总数为2 779条,其中单碱基型的数量最多,为1 473条,所占比例超过50%。这与在其他水生物种转录组报道有相似的结果。例如,岳华梅等[24]利用该测序技术对兴国红鲤 (Cyprinuscarpiovar.singuonensis)进行了SSR标记筛选,发现单碱基型占的比例最大,为47.86%。Zhou X X等[25]对刺参(Apostichopusjaponicus)进行转录组序列分析,发现单碱基型有9 154条,所占比例为75.56%。Huang Y等[26]对大鲵(Andriasdavidianus)进行转录组SSR研究,也发现单碱基型有25 100条,所占比例达到了84.3%。表明单碱基型的SSR分子标记类型可能普遍存在于水生动物中。这些数据的获得,极大地丰富了西藏山溪鲵转录本信息和基因资源,可为西藏山溪鲵相关性状的基因进行深入定位与克隆,并为后续同属物种群体遗传多样性分析与连锁图谱构建、分子标记开发、评估和保护其遗传资源、适应性进化机制等研究提供分子基础支撑。
猜你喜欢
幼儿100(2023年42期)2023-11-21 09:36:38
学与玩(2022年7期)2022-10-31 01:59:22
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
作文周刊·小学二年级版(2020年16期)2020-06-12 11:44:29
生物学通报(2019年3期)2019-02-17 18:03:58
小猕猴智力画刊(2017年12期)2017-12-27 17:30:31
小学生导刊(低年级)(2017年2期)2017-06-10 10:50:24
海峡姐妹(2017年4期)2017-05-04 04:03:51
剑南文学(2016年11期)2016-08-22 03:33:36
