
基于拉曼光谱的绞股蓝总皂苷色谱洗脱过程在线监测方法研究
2023-06-19 03:33:22谢佳丽姜新宇王青青张建兵瞿海斌
中草药 2023年12期
谢佳丽,张 胜,姜新宇,王青青,张建兵,瞿海斌*
基于拉曼光谱的绞股蓝总皂苷色谱洗脱过程在线监测方法研究
谢佳丽1,张 胜1,姜新宇2,王青青3,张建兵3,瞿海斌1*
1. 浙江大学 药物信息学研究所,浙江 杭州 310058 2. 湖南华宝通制药有限公司,湖南 长沙 410331 3. 万邦德制药集团有限公司,浙江 台州 317599
为实现绞股蓝总皂苷(saponins,GPS)色谱洗脱过程实时监测,保障纯化过程绞股蓝总皂苷质量一致性。采集色谱洗脱过程7批共计237个样本的拉曼光谱,将其中5批用于建模,2批用于外部测试,以总皂苷质量浓度、总固体量和人参皂苷Rb3(Rb3)质量浓度为指标,采用高斯过程回归(Gaussian process regression,GPR)法建立定量模型,并将GPR模型与偏最小二乘回归及支持向量机回归定量模型进行性能对比。基于拉曼光谱技术结合GPR,建立了其洗脱过程的多指标定量校正模型。总皂苷质量浓度、总固体量和Rb3质量浓度3个指标的GPR模型均具有更高的决定系数(2),训练集2均为1.00,验证集2分别为0.953、0.986、0.939,以及更低的误差均方根(root mean square error,RMSE),训练集RMSE分别为70.4、224.0、31.6 μg/mL,验证集RMSE分别为3.02、2.03、1.19 mg/mL。GPR模型在外部测试集的结果为总皂苷质量浓度、总固体量和Rb3质量浓度预测2分别达到0.947、0.954、0.837,RMSE分别为3.28、4.37、2.44 mg/mL;GPR模型能较好地反映总皂苷质量浓度和总固体量含量和变化趋势,但对Rb3质量浓度的预测能力较弱。以总皂苷质量浓度和总固体量为指标,提出的基于拉曼光谱结合GPR建模的方法可实现绞股蓝总皂苷色谱洗脱过程的实时监测。
绞股蓝;色谱洗脱;拉曼光谱;高斯过程回归;在线监测;质量一致性;总皂苷;人参皂苷Rb3;偏最小二乘回归;支持向量机;误差均方根
中药提取物的分离纯化是中药制剂生产过程中的关键环节,直接影响到产品的质量和临床疗效[1]。大孔树脂色谱常被应用于总黄酮、总皂苷、总生物碱等中药目标成分的富集、分离和纯化。但大孔树脂色谱在实际生产中缺乏过程监测方法,终点放行主要依赖于生产经验或实验室分析结果[2],耗时且滞后,难以及时反映生产状态以致生产决策延时,造成产品质量一致性较差。
绞股蓝(Thunb.) Makino是多年生草质藤本植物,为葫芦科绞股蓝属,又名“七叶胆”“五叶参”等,在临床上主要用于调节血糖、调血脂、抗肿瘤和抗血栓等,因此绞股蓝也逐渐备受关注[3-4]。绞股蓝的主要有效成分为皂苷类、多糖类、黄酮类、氨基酸类、多种维生素及微量元素等[5],其中皂苷类被视为绞股蓝中重要的标志性成分,是绞股蓝的主要药效成分[6-7]。绞股蓝总皂苷工业化生产常用的方法是大孔吸附树脂法,目前,关于绞股蓝总皂苷色谱过程在线监测方法的研究报道较少。
拉曼光谱(Raman spectroscopy)是一种快速、无损的在线检测技术,其原理是基于拉曼散射效应得到分子振动、转动相关信息从而反映化学组成,是一种常用的过程分析技术(process analytical technology,PAT)工具。拉曼光谱技术在中药[8-10]、生物药[11-13]、化学药[14-16]的生产过程已有一定的应用研究进展,通过化学计量学和机器学习等方法建立定量光谱校正模型,实现制药过程的在线监测。
主成分回归(principal components regression,PCR)[17]、偏最小二乘回归(partial least squares regression,PLS)[18-20]及支持向量机回归(support vector regression,SVR)[21-23]是常用光谱定量建模方法,但模型性能受样本数量影响显著,对于小样本数据,模型性能有时候会达不到理想效果。高斯过程回归(Gaussian process regression,GPR)是一种基于贝叶斯理论和统计学习理论的机器学习方法,适用于非线性、高维度以及小样本等复杂数据的回归问题[24-25]。
本研究以绞股蓝总皂苷色谱过程为对象,采用GPR法建立了基于拉曼光谱的在线监测方法。首先优化GPR模型的核函数,然后将建立的GPR模型与优选的PLS及SVR定量模型进行性能对比,最终将该模型应用于绞股蓝总皂苷色谱洗脱过程,为该过程智能化生产提供了研究基础。
1 仪器与材料
i-Raman Plus型便携式拉曼光谱仪,美国B&W Tek公司,配备785 nm激光源(300 mW)、电荷耦合器件检测器和BAC101型工业级拉曼探头;Agilent 1260型高效液相色谱仪,Agilent科技有限公司,配备在线脱气机、四元泵、标准型自动进样器、柱温箱和蒸发光散射检测器(evaporative light-scattering detector,ELSD);BT300-2J型流量型蠕动泵,保定兰格恒流泵有限公司;石英流通池,光程5 mm,宜兴晶科光学仪器有限公司。
人参皂苷Rb1(Rb1,批号220307,质量分数≥98%)、人参皂苷Rb3(Rb3,批号220803,质量分数≥98%)对照品均购自上海融禾医药科技有限公司。无水乙醇,分析纯,体积分数≥99.7%,上海凌峰化学试剂有限公司;纯净水,杭州娃哈哈集团有限公司。柱色谱过程中的绞股蓝上样液样品,批号T2021110101、T2021110102、T2021110103、T2021110104,由万邦德制药集团有限公司提供。
2 方法
2.1 样本采集
绞股蓝总皂苷色谱过程包含了上样、水洗、25%乙醇洗脱、70%乙醇洗脱和90%乙醇洗脱等工艺流程。为确保采样点尽可能体现洗脱过程轨迹,从70%乙醇洗脱开始计时,分时段选择不同的采样间隔来采集样本,以批次2为例:在有效成分开始被洗脱出来之前,即第5~20 min,采样间隔为5 min;在有效成分被洗脱出来至达到峰值后,即第21~30 min,采样间隔为1 min;在峰值之后至洗脱终点前,即第31~65 min,采样间隔为5 min;洗脱终点前后即第66~100 min,采样间隔为2 min;各批次实验间根据具体情况进行调整。每个样本采集时长为1 min,进行了7个批次实验,共采集了237个样本,结果见表1。
2.2 拉曼光谱采集与预处理
绞股蓝总皂苷色谱洗脱实验和光谱采集装置见图1,色谱洗脱液流出后直接进入石英流通池来采集拉曼光谱。流通池到采样口时滞约35 s,因此在光谱开始采集时需间隔35 s再采样。拉曼光谱采集参数:光谱波数范围172.91~3 201.75 cm−1,激光强度100%,积分时间800 ms,平均次数43次(由于光谱仪内部通讯和计算等原因,实验中每张光谱的实际采集时间约为1 min)。
拉曼光谱易受荧光干扰[26],以Savitsky-Golay平滑(SG,平滑点数15个)、一阶导数(first-order differential,1st)、二阶导数(second-order differential,2nd)以及标准正态变换(standard normal variate,SNV)等方法及其组合作为光谱预处理方法可以减少荧光干扰,提取有用信息。

图1 绞股蓝总皂苷色谱洗脱实验和光谱采集装置图
绞股蓝总皂苷色谱洗脱过程原始及预处理后拉曼光谱见图2。单个批次内随着70%乙醇洗脱时间增加,拉曼光谱相对强度呈现先上升后下降趋势,与含量变化趋势一致,各批次拉曼强度范围在0~65 000。光谱预处理方法通过对光谱数据平滑降噪和基线校正达到减少荧光及噪声背景干扰:SNV法降低了光谱数据中固体颗粒和样品表面散射等造成的影响;SG 1st和SG 2nd有效地消除了基线漂移及荧光噪声背景干扰,并分辨重叠峰,提高了拉曼光谱分辨率灵敏度。
2.3 绞股蓝总皂苷含量测定方法[27]
2.3.1 对照品溶液的制备 精密称取Rb1对照品6.90 mg于10 mL量瓶中,加甲醇至刻度,得质量浓度为0.690 mg/mL的对照品溶液。
2.3.2 线性关系考察 吸取对照品溶液50、100、150、200、250、300、380 μL分别置于10 mL具塞试管中,60 ℃挥干,分别加入5%香草醛冰乙酸溶液0.2 mL、高氯酸0.8 mL,混匀,密塞,置60 ℃水浴中加热15 min,取出,冷却,加冰醋酸5 mL,混匀,即质量浓度分别为34.5、69.0、104.0、138.0、172.0、207.0、262.0 μg/mL的系列对照品溶液。
以空白试剂为对照,在550 nm处测定吸光度。以质量浓度为横坐标(),吸光度为纵坐标(),绘制标准曲线,进行线性回归,得其回归方程=0.275-0.010 9,相关系数=0.999 6,线性范围34.5~262.0 μg/mL。
2.3.3 绞股蓝总皂苷定量测定方法 精密量取所采集的样本溶液适量,置10 mL具塞试管中,60 ℃挥干后,按“2.3.2”项下方法操作,测定吸光度,按照回归方程计算绞股蓝总皂苷的质量浓度。
2.4 总固体量测定方法
精密量取适量样品溶液,置已干燥至恒定质量的称量瓶中,水浴上蒸干,于105 ℃干燥3 h,移至干燥器中冷却30 min,迅速精密称定质量,计算总固体量。
2.5 Rb3含量测定方法
使用HPLC-ELSD法测定绞股蓝总皂苷色谱洗脱过程中的含量最高的皂苷成分即Rb3含量变化。
2.5.1 色谱条件 采用Agilent Zorbax SB-C18色谱柱(250 mm×4.6 mm,5 μm);流动相为乙腈-0.5%乙酸水溶液,采用梯度洗脱程序:0~25 min,5%~31%乙腈;25~33 min,31%~33.2%乙腈;33~55 min,33.2%~33.5%乙腈;55~70 min,33.5%~50%乙腈;70~81 min,50%~58%乙腈;81~91 min,58%~100%乙腈;体积流量0.8 mL/min;进样量5 μL。ELSD参数:增益100,漂移管温度80 ℃,气压0.7 MPa。色谱图见图3。

图2 绞股蓝总皂苷色谱洗脱过程原始及预处理后拉曼光谱图
2.5.2 对照品溶液的制备 精密称取Rb332.44 mg于25 mL量瓶中,加甲醇至刻度,得质量浓度为1.268 mg/mL的对照品母液;精密吸取该母液适量,用甲醇稀释25倍,制得质量浓度为51.90 μg/mL的对照品溶液。

图3 阴性对照(A)、Rb3对照品(B)和供试品溶液(C)的HPLC图
2.5.3 供试品溶液的制备 取绞股蓝总皂苷色谱70%乙醇洗脱段溶液,经0.22 μm微孔滤膜滤过,即得。缺绞股蓝阴性对照品溶液由70%乙醇溶液经0.22 μm微孔滤膜滤过而得。
2.5.4 线性关系考察 精密吸取“2.5.2”项下的对照品母液5、10、20、30 μL,以及母液稀释25倍所得的对照品溶液5、10、20 μL,按“2.5.1”项色谱条件进样检测,记录色谱峰峰面积,以Rb3对照品质量浓度对峰面积进行线性回归,得回归方程为=9.889×105-468.2,相关系数=0.999 7,线性范围0.259 5~38.04 μg/mL。
2.5.5 精密度试验 取T2021110103样品,依供试品溶液制备方法制备一份供试品溶液,依法连续进样6次,计算Rb3色谱峰峰面积的RSD为2.11%。
2.5.6 稳定性试验 取同一份T2021110103供试品溶液,于制备后0、2、4、6、12、18、24、36 h依法检测,计算Rb3色谱峰峰面积的RSD为2.46%,表明供试品溶液在36 h内稳定。

2.5.8 加样回收试验 精密称取7.16 mg Rb3对照品于10 mL量瓶中,甲醇定容,得质量浓度为0.716 mg/mL的对照品溶液。精密吸取3份527、659、791 μL质量浓度为0.716 mg/mL的对照品溶液于9个1 mL量瓶中,挥干。取9份已知质量浓度的T2021110103供试品溶液,定容至刻度,依法检测,计算Rb3低、中、高浓度平均加样回收率分别为94.76%、99.18%、94.57%,RSD分别为2.16%、2.68%、1.39%。
2.5.9 样品测定 将色谱洗脱实验所得样品,按“2.5.3”项下条件制备供试品溶液,按“2.5.1”项下色谱条件进样分析,记录峰面积,按标准曲线计算Rb3的含量。
2.6 GPR
GPR是一种基于贝叶斯方法的非参数概率模型,高斯过程(Gaussian process,GP)性质完全由均值函数和协方差函数确定,因此,GP可定义为()=GP((),(,′)),对于回归模型如下:=()+,其中,为输入变量,为观测值,是观测噪声,假设其服从高斯分布,即=(0,σ2),其中σ2是噪声的方差,因此,就可以得到的先验分布=(0,(,)+σ2I),以及观测值和预测值*的联合先验分布为
(1)
其中,*为输入待测变量,式(1)也可以表示为

利用式(2)可得到*的后验分布为
(3)


2.7 模型建立过程
将批次2~6实验样本作为建模数据集,批次1和7实验样本作为独立于建模数据集的外部测试集。利用浓度梯度法将建模样本的光谱数据划分为训练集和验证集(3∶1),由于部分样本的指标参考值缺失,各指标模型的样本集划分结果见表2。

表2 各指标模型的样本划分结果
采用GPR法建立绞股蓝总皂苷色谱洗脱过程的拉曼光谱与多个指标间的定量模型。GPR模型的性能受到核函数的影响显著,因此,建立了基于不同核函数的GPR模型,包括平方指数核(squared exponential,SE)、指数核(exponential,Exp)、有理二次核(rational quadratic,RQ)、Matern32与Matern52核。
通过贝叶斯优化找到最小化五折交叉验证损失的超参数。以相同训练集和验证集建立PLS和SVR定量模型,其中SVR模型的超参数包括惩罚参数和核参数等,通过贝叶斯优化方法以最小化五折交叉验证损失为目标来寻找最佳的超参数组合,而PLS模型同样通过最小化五折交叉验证误差均方根来优化主成分数。
模型性能评价指标包括决定系数(2)和误差均方根(root mean square error,RMSE)。将得到的GPR模型与优选的PLS以及SVR模型进行各指标预测性能的比较。
3 结果
3.1 色谱洗脱过程样本含量
以70%乙醇开始洗脱为起点,各批次样本的绞股蓝总皂苷含量、总固体量及Rb3含量的过程变化趋势见图4。可以看出,不同批次间绞股蓝总皂苷质量浓度和总固体量含量范围与变化趋势较一致,但不同批次Rb3含量存在较大差异,其原因包括上样液的Rb3含量各不相同以及各组分间竞争吸附。除此之外,批次间洗脱起点与终点存在时间差异,原因在于大孔树脂色谱柱柱效变化,批次3~7实验于大孔树脂色谱柱再生后进行,而批次1和2实验于再生前进行。
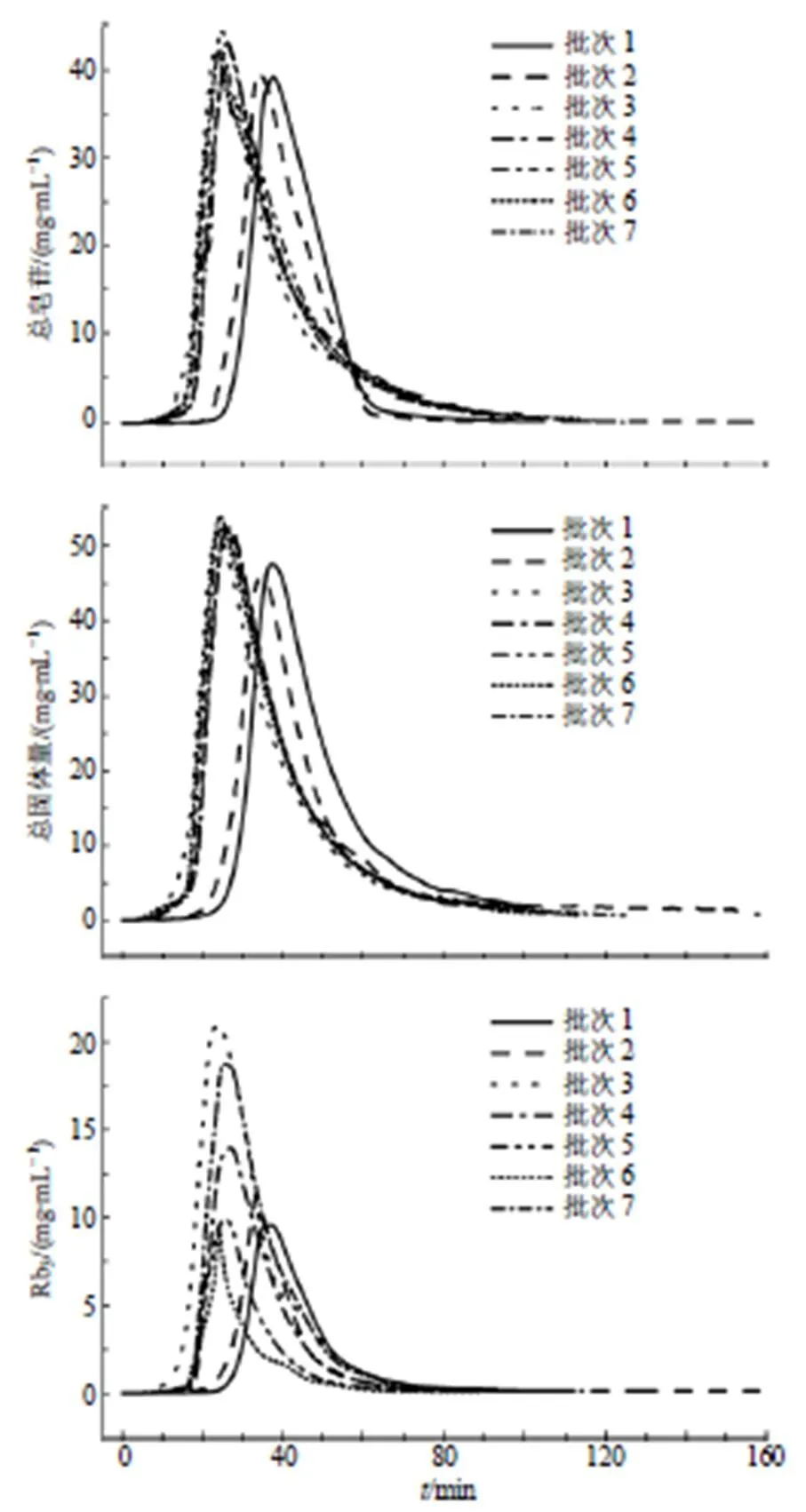
图4 绞股蓝总皂苷色谱洗脱过程的各指标变化图
3.2 不同核函数GPR模型性能比较
建立的各指标基于不同核函数的GPR模型性能对比结果见表3,预处理方法选择SG 2nd。总皂苷各GPR模型训练集的2和RMSE相差不大,观察验证集,Matern32核模型具有更高的2和更低的RMSE,总皂苷和Rb3模型同理,因此总皂苷、总固体量和Rb3的GPR模型最佳核函数均为Matern32核。优选出的3个GPR模型2都超过了0.93,表明3个模型均具有较好的拟合效果,模型性能好。
3.3 GPR、PLS和SVR模型性能比较
以相同的数据集建立并优选出PLS和SVR模型,与“3.3”项得到的GPR模型的性能比较结果见表4。总皂苷、总固体量和Rb3的PLS模型预处理方法分别为1st、1st和SG(平滑点数为15)。根据交叉验证均方根误差(root mean square error of cross validation,RMSECV)选择最佳主成分数,结果见图5。为防止过拟合,主成分数量上限设为10个,总皂苷、总固体量和Rb3的PLS模型的最佳主成分数分别为3、3和10个。SVR模型采用SNV预处理后的拉曼光谱作为输入,核函数均为线性函数。

表3 各指标基于不同核函数的GPR模型性能对比结果

表4 各模型的性能参数对比结果

图5 各指标PLS模型主成分数量选择图
观察表4可发现,总皂苷、总固体量与Rb3的GPR模型均拥有更高的2和更低的RMSE,性能最佳,其次是SVR模型,PLS模型性能最差。以验证集RMSE为评价指标,GPR总皂苷定量模型相对PLS和SVR模型预测误差分别降低了31.8%、22.7%;GPR总固体量定量模型相对PLS和SVR模型预测误差分别降低了69.5%、56.2%;GPR Rb3定量模型相对PLS和SVR模型预测误差分别降低了25.2%、37.4%。在外部测试集中,总皂苷和固含量GPR模型2均大于0.93,模型拟合效果好且具有较好的鲁棒性;Rb3的GPR模型虽然相比PLS和SVR模型性能最佳,但其2只有0.81,模型预测准测度严重下滑,说明该模型鲁棒性差。
图6为各模型预测值与参考值的相关图,可以看出,3个指标各模型中,PLS模型相关性均最差;SVR模型训练集样本均具有较好的相关性,而验证集样本相关性较差;GPR模型训练集和验证集均具有较好的相关性,说明模型拟合效果好,预测准确度高。将建立的GPR模型用于监测批次1和7绞股蓝总皂苷色谱洗脱过程的总皂苷质量浓度、总固体量和Rb3质量浓度,结果如图7所示。GPR模型能较好地预测总皂苷质量浓度和总固体量,而对Rb3质量浓度预测准确度较差。其原因可能在于Rb3含量在各上样液中差异较大,且Rb3与其他各成分存在竞争吸附,而本实验中采集的样本较少,不足以充分反映这些情况,导致GPR模型对Rb3质量浓度的预测性能较差。因此,总皂苷质量浓度和总固体量更适合作为绞股蓝总皂苷色谱洗脱过程的监测指标,通过GPR模型实现色谱洗脱过程的可视化,帮助实验者判断收集起点和洗脱终点。

图6 各模型预测值与参考值的相关图

图7 绞股蓝总皂苷色谱洗脱过程中总皂苷质量浓度、总固体量和Rb3质量浓度实时监测
4 讨论
本研究建立了基于拉曼光谱技术的绞股蓝总皂苷色谱洗脱过程的总皂苷浓度、总固体量和Rb3质量浓度实时监测方法。结果表明,该方法能精准地预测绞股蓝总皂苷色谱洗脱过程的总皂苷浓度和总固体量含量以及变化趋势,但对Rb3的预测能力相对较弱。后续研究中可通过增加训练集代表性样本数量、筛选特征波长范围、更改样本集划分方法及建模算法等手段提高光谱信息挖掘能力,进一步提高模型对Rb3的预测能力。以总皂苷浓度和总固体量为指标,该方法有助于生产人员判断绞股蓝总皂苷洗脱起点和终点,可将该思路应用到绞股蓝总皂苷色谱洗脱上样过程,指导上样吸附时间的调整优化,以保障绞股蓝总皂苷的质量一致性。
值得注意的是,相比PLS定量模型,SVR和GPR模型出现了过拟合现象。模型过拟合根本原因在于模型对数据变异的高解释力并不等同于高预测力。数据变异包括来源自变量的变异和来源于抽样随机误差的变异,模型拟合度即对数据变异的总解释度。SVR和GPR法相比PLS增加了模型复杂度,提高了模型拟合度,解释了更多的数据变异,但过度拟合反而偏离了数据生成的真实过程,导致对非样本数据预测力降低。后续研究中可采用正则化和增大样本量等来降低过拟合。
将拉曼光谱分析技术应用于中药领域,最主要的限制首先是大部分企业现有设备难以满足PAT工业应用需求,需要对生产设备进行改造以及配置PAT工具,前期成本投入大;其次实际生产过程存在颗粒及气泡问题,导致光谱采集不稳定,影响模型预测结果;最后是监管难题,国内对于PAT相关的法规和指导文件较为缺乏,在工艺变更等审批上存在挑战,企业对于实施PAT具有诸多顾虑。
利益冲突 所有作者均声明不存在利益冲突
[1] 杨敏, 张天锡, 史磊, 等. 大孔吸附树脂分离纯化中药成分影响因素探讨 [J]. 中草药, 2020, 51(15): 4050-4058.
[2] 范冬冬, 匡艳辉, 董利华, 等. 基于“伴随标志物”在线控制技术的绞股蓝总皂苷纯化工艺及成分鉴定研究 [J]. 中国中药杂志, 2017, 42(7): 1331-1337.
[3] 白宏, 王亚, 葛维娟, 等. 绞股蓝皂苷的抗癌作用机制研究进展 [J]. 西北药学杂志, 2019, 34(4): 564-567.
[4] 邓芙蓉, 王翰林, 谢佩佩, 等. 绞股蓝皂苷对人胃癌细胞增殖和凋亡的影响及作用机制 [J]. 中国药理学通报, 2023, 39(4): 646-652.
[5] 张欣怡, 夏明明. 绞股蓝化学成分的降血脂机制研究进展 [J]. 光明中医, 2020, 35(8): 1271-1274.
[6] 杜晓鸿. 绞股蓝发酵口服液的制备及对小鼠免疫功能的影响 [D]. 重庆: 西南大学, 2022.
[7] 史琳, 王志成, 时圣明, 等. 绞股蓝皂苷水解产物化学成分和药理作用研究进展[J]. 药物评价研究, 2017, 40(5): 711-716.
[8] 殷文俊, 唐建飞, 郑洁, 等. 基于拉曼光谱实时监测甘草配方颗粒的提取过程 [J]. 中草药, 2021, 52(18): 5560-5568.
[9] Ru C L, Wen W, Zhong Y. Raman spectroscopy for on-line monitoring of botanical extraction process using convolutional neural network with background subtraction [J]., 2023, 284: 121494.
[10] Tao Y, Bao J Q, Liu Q,. Application of deep-learning algorithm driven intelligent Raman spectroscopy methodology to quality control in the manufacturing process of Guanxinning Tablets [J]., 2022, 27(20): 6969.
[11] Gibbons L, Rafferty C, Robinson K,. Raman based chemometric model development for glycation and glycosylation real time monitoring in a manufacturing scale CHO cell bioreactor process [J]., 2022, 38(2): e3223.
[12] Westley C, Fisk H, Xu Y,. Real-time monitoring of enzyme-catalysed reactions using deep UV resonance raman spectroscopy [J]., 2017, 23(29): 6983-6987.
[13] Hara R, Kobayashi W, Yamanaka H,. Development of Raman calibration model without culture data for in-line analysis of metabolites in cell culture media [J]., 2023: 521-533.
[14] 刘兰玲. 近红外与拉曼光谱技术在硝苯地平原辅料及其溶出过程中的应用研究 [D]. 济南: 山东大学, 2021.
[15] Inoue M, Osada T, Hisada H,. Quantitative monitoring of cocrystal polymorphisms in model tablets using transmission low-frequency Raman spectroscopy [J]., 2023, 112(1): 225-229.
[16] Zhu Q X, Li X H, Li D,. A rapid therapeutic drug monitoring strategy of carbamazepine in serum by using coffee-ring effect assisted surface-enhanced Raman spectroscopy [J]., 2022, 28(1): 128.
[17] 孟庆龙, 尚静, 黄人帅, 等. 基于主成分回归的猕猴桃可溶性固形物无损检测 [J]. 包装工程, 2021, 42(3): 19-24.
[18] 阎续, 沈丽娟, 胥文彦, 等. 拉曼光谱用于CHO细胞培养液多指标快速分析 [J]. 高校化学工程学报, 2019, 33(4): 872-877.
[19] He H J, Wang Y L, Ou X Q,. Rapid determination of chemical compositions in chicken flesh by mining hyperspectral data [J]., 2023, 116: 105069.
[20] Teye E, Amuah C L Y, Yeh T S,. Nondestructive detection of moisture content in palm oil by using portable vibrational spectroscopy and optimal prediction algorithms [J]., 2023, 2023: 3364720.
[21] 韩斯琴高娃, 李楠, 薛兰, 等. 拉曼光谱技术结合主成分分析-支持向量机对砷类矿物药的分类识别研究 [J]. 分析科学学报, 2022, 38(2): 224-228.
[22] Ikram R M A, Mostafa R R, Chen Z H,. Advanced hybrid metaheuristic machine learning models application for reference crop evapotranspiration prediction [J]., 2022, 13(1): 98.
[23] Wang C Z, Li M Y, Yan J P. Forecasting carbon dioxide emissions: Application of a novel two-stage procedure based on machine learning models [J]., 2023, 14(2): 477-493.
[24] 姚煜, 胡涛, 付建勋, 等. 小样本分散数据的回归建模和多目标优化 [J]. 上海大学学报: 自然科学版, 2022, 28(3): 451-462.
[25] Wu S J, Cui T C, Li Z,. Real-time monitoring of the column chromatographic process ofpart I: End-point determination based on near-infrared spectroscopy combined with machine learning [J]., 2022, 46(19): 9085-9097.
[26] Goldrick S, Lovett D, Montague G,. Influence of incident wavelength and detector material selection on fluorescence in the application of Raman spectroscopy to a fungal fermentation process [J].(Basel, Switzerland), 2018, 5(4): 79.
[27] 马泽刚, 黄春花, 钟辉云, 等. 八个不同产地绞股蓝总皂苷含量及抗氧化活性测定[J]. 湖北农业科学, 2018, 57(14): 109-113.
On-line monitoring method of chromatographic process ofsaponins based on Raman spectroscopy
XIE Jia-li1, ZHANG Sheng1, JIANG Xin-yu2, WANG Qing-qing3, ZHANG Jian-bing3, QU Hai-bin1
1. Pharmaceutical Informatics Institute, College of Pharmaceutical Sciences, Zhejiang University, Hangzhou 310058, China 2. Hunan Huabaotong Pharmaceutical Co., Ltd., Changsha 410331, China 3. Wanbangde Pharmaceutical Group Co., Ltd., Taizhou 317599, China
In order to realize the real-time monitoring of chromatographic process ofsaponins (GPS) and ensure the quality uniformity and batch consistency.The Raman spectra of 237 samples collected in seven batches during the chromatographic process, of which five batches were used as modeling sets and two batches were used as external test sets. With total saponin concentration, total solids and ginsenoside Rb3concentration as indexes, Gaussian process regression (GPR) method was used to establish the model, and the performance was compared with partial least squares and support vector machine regression quantitative models, and the method was applied to external test sets for validation.Multi-index quantitative correction models were established based on Raman spectroscopy combined with GPR. The results showed that the GPR models of the three indexes had higher coefficient of determination (2) and lower root mean square error (RMSE). The2of the training sets were all 1.00, and the2of the verification sets were 0.953, 0.986, and 0.939, respectively. The RMSE of the training sets were 70.4, 224.0, 31.6 μg/mL, and the RMSE of the verification sets were 3.02, 2.03, 1.19 mg/mL, respectively. The results of external test sets showed that the prediction2of total saponin concentration, total solid content and ginsenoside Rb3concentration were 0.947, 0.954 and 0.837, respectively, and RMSE were 3.28, 4.37 and 2.44 mg/mL, respectively. GPR model can predict the content and trend of total saponin and total solid well, but it is weak in predicting ginsenoside Rb3concentration.With total saponins concentration and total solids as indexes, this method can realize the real-time monitoring of the chromatographic process of GPS.
; chromatographic process; raman spectrum; gaussian process regression; on line monitoring; quality conformance; total saponins; ginsenoside Rb3; partial least squares; support vector machine regression
R283.6
A
0253 - 2670(2023)12 - 3824 - 10
10.7501/j.issn.0253-2670.2023.12.009
2023-01-11
国家中医药管理局“组分中药与智能制药多学科交叉创新团队”(ZYYCXTD-D-2020002)
谢佳丽(1998—),女,硕士研究生,研究方向为新药创制工程。E-mail: 1547670486@qq.com
通信作者:瞿海斌,博士生导师,从事制药过程质量控制研究。Tel: (0571)88208428 E-mail: quhb@zju.edu.cn
[责任编辑 郑礼胜]
猜你喜欢
昆明医科大学学报(2021年6期)2021-07-31 07:39:56
中成药(2018年6期)2018-07-11 03:01:10
中成药(2018年1期)2018-02-02 07:20:05
恋爱婚姻家庭·养生版(2017年12期)2017-12-07 22:52:44
中成药(2017年8期)2017-11-22 03:18:44
恋爱婚姻家庭(2017年36期)2017-07-22 14:33:35
陕西画报(2016年1期)2016-12-01 05:35:31
特产研究(2016年3期)2016-04-12 07:16:17
郑州大学学报(理学版)(2013年3期)2013-03-11 20:30:38
化学分析计量(2013年1期)2013-03-11 16:37:15
