
广州市花都地区罕见珠蛋白生成障碍性贫血基因型分布及血液学特征*
2023-06-15 07:48:42鞠爱萍付晓彤刘艳霞许碧秋陈武玲孟祥荣
检验医学与临床 2023年11期
鞠爱萍,付晓彤,刘艳霞,林 铿,许碧秋,陈武玲,孟祥荣
1.广东省广州市花都区妇幼保健院检验科,广东广州 510800;2.南昌大学玛丽女王学院,江西南昌 330031
珠蛋白生成障碍性贫血又称为地中海贫血,简称地贫,是一种常染色体单基因隐性遗传病,是一组世界上最常见的慢性溶血性贫血病,主要以α、β-地贫最为常见[1]。当前国内市场上的地贫诊断试剂盒提供的检测范围主要是针对我国南方常见的6种α-地贫和17种β-地贫的基因类型,检测范围涵盖了我国人群中95%~98%的α、β-地贫基因型,在一定程度上会造成2%~5%的罕见或未知突变地贫基因型漏诊。随着分子检测技术的不断发展,罕见或未知突变地贫基因型不断被发现。广州市花都地区是地贫高发区,对常规α、β-地贫的基因型及血液学表型已有相关文献报道[2-3],但对罕见地贫的研究尚处于初期阶段,基于此,本研究以血液学特征为基础,开展了罕见地贫基因型检测,调查了广州市花都地区人群罕见地贫基因型的分布及其血液学表型特征,探讨了该地区罕见地贫人群携带率,阐述了罕见地贫基因型与表型的关系,不仅可完善该地区地贫基因突变谱,还可为地贫产前咨询和防控提供参考依据。
1 资料与方法
1.1一般资料 选择2017年1月至 2022年6月在广州市花都区妇幼保健院进行地贫基因检测者33 014例患者作为研究对象。罕见α-地贫纳入标准:平均红细胞体积(MCV)<82 fL和(或)平均红细胞血红蛋白含量(MCH)<27 pg、血红蛋白(Hb)A2<2.5%,但常见地贫基因型为阴性或基因型与血液学表型不一致者或已生过重型α-地贫患儿但未找到原因者。罕见β-地贫纳入标准:MCV<82 fL和(或)MCH<27 pg、且HbA2≥3.5%或(和)HbF≥5.0%,常规地贫基因型为阴性或中重型β-地贫表型,但常规基因型为杂合子者。排除标准:缺铁性贫血、铅中毒、甲状腺功能亢进症、严重感染者及其他类型的Hb病者。所有患者均了解本研究并签署知情同意书。本研究经花都区妇幼保健院伦理委员会审批通过。
1.2试剂与仪器 选用Sysmex XN-1000全自动血液分析仪(日本Sysmex公司)、ABI2720扩增仪(美国ABI公司)、Capillary2全自动毛细管电泳仪(法国SEBIA公司)、HB-2012A医用核酸分子快速杂交仪(广东潮州凯普公司)、YY-6C型电泳仪(北京六一仪器厂)、ABI 3500 DNA测序仪(美国ABI公司)等设备。常规α、β-地贫基因检测试剂盒均购自广东潮州凯普公司。
1.3方法
1.3.1血液学参数水平测定 采集受检者外周血2 mL,乙二胺四乙酸二钾抗凝,颠倒混匀,充分抗凝,每批次实验均处于在控的状态时采用Sysmex XN-1000全自动血液分析仪进行MCV(参考范围:82~100 fL)、MCH(参考范围:27~34 pg)、Hb(参考范围:男120~180 g/L,女110~170 g/L)等血液学参数水平测定;使用Capillary2全自动毛细管电泳仪进行受检者HbA(参考范围:94.5%~97.5%)、HbA2(参考范围:2.5%~3.5%)、HbF(参考范围:0%~2.5%)及其他异常Hb等检测。
1.3.2常见地贫基因型检测 采用跨越断裂点聚合酶链反应(Gap-PCR)联合导流杂交法检测6种常见α-地贫的基因型,即--SEA、-α3.7、-α4.2、αCSα、αQSα和αWSα,以及17种常见β-地贫基因型,即IVS-Ⅱ-654(C-T)、βE(G-A)、CD41-42(-TCTT)、-28(A-G)、-29(A-G)、CD14-15(+G)、CD71-72(+A)、CD27-28(+C)、-30(T→C)、CD17(A-T)、IVS-Ⅰ-1(G-T、G-A)、IVS-Ⅰ-5(G-C)、Int(T-G)、CD43(G-T)、CD31(-C)、-32(C→A)和CAP(A-C或-AAAC)。
1.3.3罕见地贫基因型检测 (1)罕见α-地贫基因型检测。Gap-PCR联合导流杂交法检测6种常见α-地贫基因型为阴性时,采用Gap-PCR检测罕见缺失性α-地贫基因型[泰国型地贫(--THAI)和菲律宾型--FIL]也为阴性时,再采用多重连接探针扩增技术(MLPA)检测未知α-缺失型地贫,Sanger测序分析α-珠蛋白基因罕见点突变;凯普地贫检测膜条出现4.2浅点时进行α-地贫融合基因检测;正常对照点、SEA、3.7或4.2三点同时出现时则进行α-地贫香港型地贫基因(HKαα)检测。(2)罕见β-地贫基因型检测。反向点杂交技术检测常见 β-地贫点突变为阴性时,采用Gap-PCR检测两种缺失型β-地贫基因型[中国型Gγ+(Aγδβ)0和东南亚型遗传性持续性胎儿Hb增高症(SEA-HPFH)]也为阴性时,再采用Sanger测序分析β-珠蛋白基因新发点突变或采用MLPA检测未知β-缺失型地贫。在HbVar数据库中查找罕见和未知的突变位点。
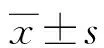
2 结 果
2.1罕见地贫基因型检出情况 33 014例受检者中检测出疑似罕见地贫基因型突变样本188例,年龄1个月至55岁。最终诊断为罕见地贫基因型94例,其中罕见α-地贫基因型46例,罕见β-地贫基因型48例。罕见地贫基因型检出率为0.28%(94/33 014),罕见α-地贫基因型检出率为0.14%(46/33 014),罕见β-地贫基因型检出率为0.15%(48/33 014)。
2.2罕见α-地贫基因型分布及血液学特征 罕见α-地贫基因型共有11种突变类型,α基因缺失型和融合型37例4种类型:1例HKαα/αα、7例--SEA/HKαα、3例-α4.2/HKαα及1例合并β17/βN、4例--THAI/αα;20例 Fusion gene/αα;1例αααanti4.2。α-地贫突变型9例7种类型:2例CD 40、2例3′UTR +71 G>C、2例IVS Ⅱ-55 T>G、1例IVS Ⅱ-119-126-CTCGGCCC>+G)、1例IVS Ⅱ-119 G>CTCGGCCC、1例CD 124 TCC>ACC、1例IVS Ⅱ-107G>C。基因型HKαα/αα、αααanti4.2、3′UTR +71 G>C及其复合-α3.7/αα、IVS Ⅱ-119 G>CTCGGCCC复合IVS Ⅱ-55 T>G复合-α4.2/αα的MCV、MCH正常,HbA2正常或稍低,而Fusion gene/αα的MCV、MCH处于临界值、HbA2正常。其余类型的MCV、MCH均降低,HbA2正常或降低,在复合β-地贫时HbA2则升高。见表1、2。

表1 罕见缺失型和融合型α-地贫基因型分布及其血液学特征
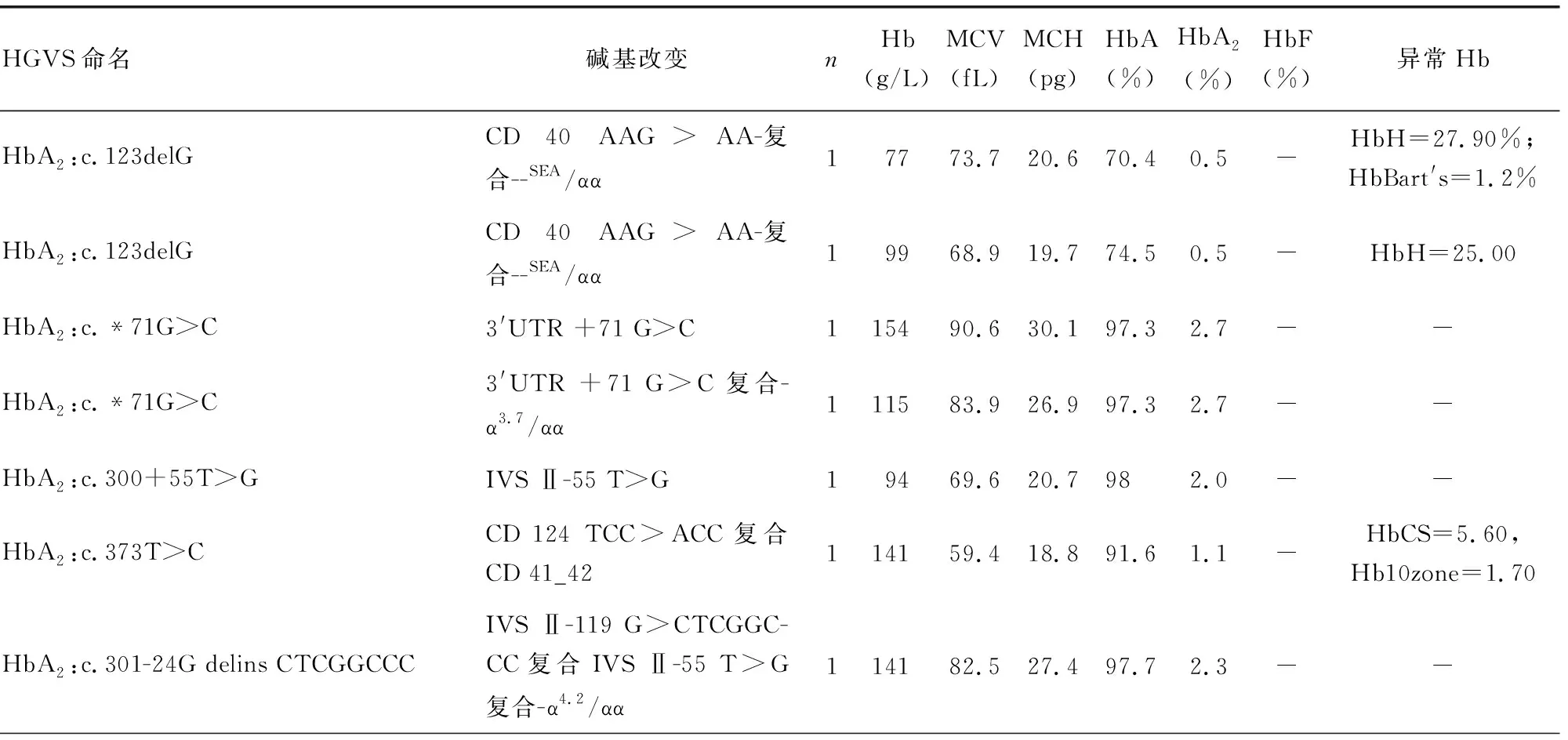
表2 罕见突变型α-地贫基因型分布及其血液学特征
2.3罕见β-地贫基因型分布及血液学特征 罕见β-地贫基因型共8种突变类型,β-基因缺失型37例2种类型,分别为10例中国型Gγ+(Aγδβ)0,27例SEA-HPFH;β-基因突变型11例6种类型,6例-90、CD37、IVS Ⅱ-2-T、IVS Ⅱ-132、IVS Ⅱ-300-308、IVS Ⅱ-667 各1例。中国型Gγ+(Aγδβ)0和SEA-HPFH杂合子及复合--SEA/αα的MCV、MCH均降低,HbF均升高,中国型Gγ+(Aγδβ)0的HbA2正常,而SEA-HPFH的HbA2升高。β-点突变单纯杂合子的MCV、MCH均降低,除IVS Ⅱ-300-308、IVS Ⅱ-667 T>C的HbA2正常或稍低外,其他类型均升高。见表3、4。
表3 罕见缺失型β-地贫基因型分布及其血液学特征
表4 罕见突变型β-地贫基因型分布及其血液学特征
3 讨 论
本研究共发现罕见α-地贫基因型46例,罕见β-地贫基因型48例,表明罕见α、β变异携带者在该地区人群中占有一定的比例,罕见地贫基因型检出率为0.28%(94/33 014),罕见α-地贫基因型检出率为0.14%(46/33 014),罕见β-地贫基因型检出率为0.15%(48/33 014),罕见基因型的携带主要以杂合子为主,其中α基因变异占48.94%(46/94),β基因变异占51.06%(48/94)。α、β点突变在外显子和内含子区域均可发生,α点突变多数发生在功能较强的α2基因上;β点突变可发生在编码区,也可以发生在5′和3′端的非编码区,部分点突变可引起相应的血液学表型变化。
HKαα是一种较为罕见的α-地贫基因型,采用凯普试剂盒Gap-PCR检测时结果显示为-α3.7/αα,而真实结果可能为-α3.7/αα、-α3.7/HKαα或HKαα/αα。有研究表明,福建省人群中HKαα检出率为0.36%,HKαα在-α3.7阳性标本中的检出率为7.27%[4]。本研究在检测膜条正常对照点、SEA、3.7或4.2三点同时出现时才进行α-地贫HKαα检测,并没有针对检测结果为-α3.7/αα的样本进行HKαα检测。据报道,广东地区HKαα/αα在-α3.7阳性标本中的检出率为4.31%[5],说明本研究报道应该存在一定比例的漏诊。本研究共检测出12例HKαα,检出率为0.04%(12/33 014),其真实的检出率应该高于0.04%,其中检测出7例--SEA/HKαα,检出率为0.02%(7/33 014),低于相关研究地贫--SEA/HKαα的检测率(0.03%)[6];--SEA/HKαα属于轻型地贫,临床症状与--SEA/αα相似,但明显轻于--SEA/-α3.7,故当夫妇双方一方为HKαα/αα、另一方为--SEA/αα时不属于地贫高风险家庭,可以不用做产前诊断[7]。本研究还检出3例-α4.2/HKαα,其MCV、MCH处于正常范围,无贫血症状,属于静止型地贫,而-α4.2/HKαα复合β17/βN的MCV、MCH明显降低,HbA2升高,表现为β-地贫的血液学表型,原因应该是由于HKαα合并β-地贫时加剧了α、β的不平衡所致。
--THAI为α-蛋白基因簇的大片段缺失,在我国南方是仅次于常规3种缺失型的地贫。有研究报道,在广西壮族自治区梧州市1 365对地贫筛查双阳受检者中检出8例--THAI,检出率为0.59%[8]。有研究在1 543对夫妇中检出7例,检出率为0.45%[9],当合并其他类型α地贫基因型时也会产生中、重型地贫。现已有--THAI合并--SEA产生重型地贫胎儿的文献报道,而且发生水肿症状更早[10]。本研究检测出4例,检出率为0.01%(4/33 104),其中1例女方地贫基因型为--THAI/αα复合SEA-HPHF/βN,男方地贫基因型为-α3.7/αα复合βN/βN,这对夫妇应纳入地贫高风险人群,怀孕后应及时做产前诊断,以防生育--THAI/-α3.7中间型地贫患儿。
α-融合基因最先是在2013年1例家系分析中被检出,据文献报道,α-地贫融合基因合并--SEA/时可引起HbH病[10]。夫妇一方为α-地贫融合基因、另外一方为--SEA/αα地贫时可能会有生育HbH病患儿的风险,应该做产前诊断[11]。在2019年海南黎族人群中1例家系的文献报道中融合基因杂合子呈小细胞低色素性贫血,MCV、MCH明显降低,轻度贫血[12]。2020年有学者报道了采用多色探针熔解曲线方法检测融合基因,并用一代测序技术进行了验证[13]。2021年本研究团队报道了广州市花都地区融合基因检测方法及应用评价,详细介绍了采用Gap-PCR对融合基因的筛查及应用评价[14]。本研究共检测出20例Fusion gene/αα,检出率为0.06%(20/33 104),MCV和MCH均在临界范围,携带者临床表型正常,与相关报道有所不同[12],说明融合基因具有明显的人群和地域差异,与广州市花都地区以本地客家人群为主有关,是否为客家人常规携带的地贫基因型尚有待于进一步考究。
本研究缺失型罕见β-地贫包括中国型Gγ+(Aγδβ)0和SEA-HPFH两种基因类型,二者均以HbF高为特点,杂合子一般无贫血表现,由于发病机制不同,SEA-HPFH临床表型要轻于中国型Gγ+(Aγδβ)0,当合并常见β-地贫时可产生中、重型临床表型。所以,当夫妇双方一方为缺失型β-地贫、另一方为常见β-地贫时要做产前诊断[15]。本研究检测出中国型Gγ+(Aγδβ)0杂合子9例,SEA-HPFH杂合子24例,1例SEA-HPFH/β-28,HbF高达77.40%,HbA2为3.80%;有文献报道,中国型Gγ+(Aγδβ)0复合βE,HbF高达54.1%,HbA2为2.5%[16]。以上说明HbF可以代替HbA行使功能,在合并不同常见β-地贫基因型时血液学表型不同。-90位点最早是在葡萄牙人群中被发现[17],本研究检出6例HBB:c.-140C>T (-90 C>T)位点的地贫,3例为杂合子突变,1例为合并α1基因HBA1:c.301-24CTCGGCCCdelinsG(IVS Ⅱ-119-126-CTCGGCCC>+G)位点突变的杂合子,1例合并-α3.7/αα,1例合并-α4.2/αα,单纯的杂合子突变为小细胞低色素性血液学表型,有少量的HbF存在,HbA2≥3.5%,是轻型β-地贫的表现,与相关报道结论一致[18]。
综上所述,广州市花都地区罕见地贫基因型复杂多样,具有独特的血液学特性,突变在外显子和内含子、编码区和非编码区均有分布。对广州市花都地区出现的相对高频的特有地贫基因型,如-90、融合基因应该有针对性地纳入广州市花都地区免费地贫筛查范围;在临床工作中一定要坚持基因型和血液学表型相一致的原则,并结合受检者临床症状、不良孕产史及B超影像学检查等综合分析,使用其他先进技术进一步检测以确诊,防止漏检,以此发现更多的罕见地贫基因型及新发突变,对产前地贫咨询和地贫防控措施的完善具有指导作用。
猜你喜欢
中国新闻周刊(2023年43期)2023-12-13 09:56:40
中国毕业后医学教育(2022年1期)2022-08-19 02:51:34
房地产导刊(2020年9期)2020-10-28 08:37:26
东坡赤壁诗词(2020年3期)2020-07-04 02:50:05
文苑(2020年5期)2020-06-16 03:18:30
莫愁(2019年34期)2020-01-01 02:18:10
伴侣(2019年7期)2019-07-25 06:34:47
莫愁·智慧女性(2019年12期)2019-06-01 10:12:59
湖北农业科学(2015年11期)2015-07-31 08:21:47
实验动物与比较医学(2014年3期)2014-02-28 14:52:55
