
利用shRNA慢病毒高效敲降雄性小鼠睾丸基因
2023-06-15 02:14龚洁卜治文徐晨叶岚谢婕
南京医科大学学报(自然科学版) 2023年6期
龚洁,卜治文,徐晨,叶岚,谢婕
南京医科大学生殖医学国家重点实验室,江苏 南京 211166
小鼠的精子发生是一个高度复杂且有序的过程,涉及多个与精子发生相关的基因[1-3]。不同的精子发生相关基因在不同的时间窗内准确而有序地表达是精子发生成功的必要条件。为了更好地研究这些基因在精子发生过程中发挥何种功能,需要研究者建立相关基因的基因敲除动物模型。近年来,有研究者基于RNA 干扰(RNA interference,RNAi)现象开发基因敲减平台,通过构建pSliencer-GFP-shRNA 表达载体,并递送至小鼠睾丸内,成功敲降目的基因的表达[4]。
RNAi 是指小干扰RNA(small interfering RNA,siRNA)诱发导致的同源mRNA高效特异性降解,从而引起基因表达沉默的现象[5]。siRNA结合至RNA诱导的沉默复合物(RNA-induced silencing complex,RISC)[6-7],再释放siRNA 的随从链从而激活RISC。激活后的RISC在siRNA的引导链的指导下降解其同源互补的mRNA,从而沉默特定基因的表达[8]。shRNA(short hairpin RNA)载体在细胞核内转录为pri-shRNA(primary shRNA),然后在双链RNA结合蛋白DGCR8和Drosha 蛋白的加工下形成pre-shRNA(precursor shRNA)[9]。pre-shRNA 在Exportin-5 等蛋白因子的协助下从细胞核转运至细胞质[10]。细胞质中的pre-shRNA在Dicer酶等蛋白因子加工下去除其茎环结构成为siRNA,从而启动RNAi过程来沉默目的基因表达[11]。基于这一原理,有研究者设计靶向特定序列的shRNA敲降载体,从而诱导RNA特异性降解[12]。
传统的基因敲除动物模型一般使用CRISPR/Cas9 等基因编辑技术进行遗传学模型的构建[13]。即使CRISPR/Cas9 技术经过多年发展已经趋于成熟,但仍存在耗时较长、费用较高、不能在特定时间点敲除目的基因,以及脱靶效应等诸多问题[14]。同时,某些基因的敲除小鼠模型存在胚胎死亡现象,常导致实验人员无法得到敲除小鼠。为了规避这些问题,需要研究者开发一个快速、特异、安全的睾丸原位基因敲减平台。近些年来,很多研究通过向睾丸递送siRNA[15]或反义寡核苷酸(antisense oligonucleotides,ASO)[16]来完成睾丸特定基因的敲减。虽然向小鼠睾丸递送siRNA 和ASO 可以规避CRISPR/Cas9等基因技术存在的问题,但也存在着作用时间维持较短的问题[17]。shRNA敲降载体具有表达shRNA的元件,可以通过慢病毒整合到基因组上稳定表达。研究者可以向小鼠睾丸递送shRNA慢病毒敲降载体基因来维持稳定、长效的基因表达沉默。本研究通过构建靶向Sox30基因的shRNA慢病毒敲降载体,通过网微注射进入出生24 d小鼠睾丸来敲降Sox30基因转录本,观察Sox30蛋白是否可被持续敲降,及生精管腔中生殖细胞是否有发育异常现象。
1 材料和方法
1.1 材料
实验所使用的小鼠均为24 日龄C57BL/6J(SPF级)雄性小鼠(南京医科大学实验动物中心),本研究经南京医科大学实验动物福利伦理委员会批准。Oligo(上海生工公司);限制性内切酶BglⅡ、HindⅢ、XbaⅠ、XhoⅠ、CutSmart Buffer、10×NEB Buffer3.1(NEB 公司,美国);FastPure Gel DNA Extraction Mini Kit(南 京Vazyme公司);EndoFree Mini Plasmid Kit Ⅱ(TIANGEN公司,美国);胰酶、胎牛血清、DMEM(Gibco 公司,美国);琼脂糖(上海生工公司),TRIzol(Thermo Fisher Scientific 公司,美国);RT reagent Kit(TaKaRa 公司,日本);DAPI(Sigma 公司,美国);花生凝集素(peanut agglutinin,PNA)(Vector Labs公司,美国),EGFP抗体、Sox30抗体、二抗(Abclonal公司,美国)。
1.2 方法
1.2.1 Oligos退火形成双链
将公司合成的shSox30-oligo1 和shSox30-oligo2用高压灭菌后的ddH2O溶解。按照4 μL oligo1、4 μL oligo2、2 μL 10×NEB buffer2、10 μL ddH2O配置退火体系。95 ℃水浴退火5 min,自然冷却至室温,4 ℃保存。
1.2.2 载体线性化
空载环状的pSilencer 载体按照2 μg pSilencer载体、1 μLXbaⅠ内切酶、1 μLXhoⅠ内切酶、5 μL CutSmart Buffer,再加ddH2O 补齐至50 μL体系,37 ℃水浴3 h。将酶切后的产物在1%琼脂糖凝胶中电泳30 min,使用DNA凝胶回收试剂盒回收目的产物。空载的pSuper载体分两步进行酶切,先按照2 μg pSuper 载体、1 μLBglⅡ内切酶、5 μL 10×NEB Buffer 3.1,加ddH2O 补齐至50 μL 体系,37 ℃水浴3 h。线性产物使用试剂盒纯化后,再按照2 μg pSuper线性载体、1 μLHindⅢ内切酶、5 μL 10×NEB Buffer2.1,加ddH2O 补齐至50 μL 体系,37 ℃水浴3 h。1%琼脂糖凝胶中电泳30 min,使用DNA凝胶回收试剂盒纯化产物得到双酶切的线性pSuper产物。同样地,pSuper-shSox30按照pSuper酶切体系酶切。
1.2.3 pSuper-shSox30 与pSliencer-GFP-shSox30 的连接与鉴定
20 ng线性pSuper载体、1 μL 10×T4 Buffer、1 μLT4 DNALigase、再加双链Oligos 补足至10 μL 体系,16 ℃过夜连接。连接产物转化,涂氨苄抗性平板,挑单克隆菌落,经菌落PCR后初步选取阳性菌落送测序验证。测序验证成功连接的pSuper-shSox30,酶切(同1.2.2)回收小片段,与线性pSilencer按照20 ng pSilencer、20 ng pSupersh-shSox30 小片段、1 μL 10×T4 Buffer、1 μL T4 DNA Ligase、ddH2O 补至10 μL,16 ℃过夜连接。连接产物转化,挑取单克隆菌落测序验证。

表1 shSox30核苷酸序列Table 1 The oligonucleotide sequences of shSox30
1.2.4 293T细胞培养与转染
293T 细胞用含10%胎牛血清、100 U/mL 青霉素、100 U/mL 链霉素的DMEM 培养基于37 ℃、5%CO2的恒温培养箱常规培养,每1~2 d换液1次,当细胞密度达70%~80%时转染。按照2.5 μg 质粒、50 μL 0.5 mmol/L CaCl2、100 μL 2×HBS,加水补齐至200 μL配置转染试剂。转染后36~48 h收样。空白组为未转染质粒组,过表达组为单转Sox30 过表达质粒PRK5-HA-Sox30,对照组为Sox30 过表达质粒PRK5-HA-Sox30 与shScramble 共转染,实验组为Sox30过表达质粒PRK5-HA-Sox30分别与shSox30-1、shSox30-2共转染。
1.2.5 RNA提取与RT-qPCR
细胞加入TRIzol试剂,冰上研磨至组织充分裂解,12 000 r/min 4 ℃离心30 min,取上清并加入200 μL氯仿,剧烈摇晃后室温静置3 min,12 000 r/min 4 ℃离心30 min,取上清并加入等体积的异丙醇,静置10 min 后12 000 r/min 4 ℃离心15 min,75%乙醇清洗沉淀后加入适量DEPC水溶解RNA。通过紫外可见分光光度计检测RNA浓度和纯度;采用TaKaRa的PrimeScript RT reagent Kit逆转录试剂盒合成cDNA,qPCR反应程序:95 ℃30 s,95 ℃5 s,60 ℃30 s。
1.2.6 慢病毒包装与网微注射
病毒上清溶液在4 ℃、50 000g转速下离心90 min以获得高滴度的慢病毒。再使用高滴度的慢病毒包装pSilencer-GFP-shSox30 敲降载体形成慢病毒敲降载体。慢病毒敲降载体与台盼蓝染液混合后使用毛细管注射进入出生24 d 的小鼠睾丸生精小管。
1.2.7 免疫荧光检测
TBS清洗组织切片后用1%TBST配制的含10%鸡血清的封闭液室温封闭1 h,一抗稀释液(EGPF抗体1∶500)4 ℃过夜孵育,加入二抗稀释液(PNA+绿色荧光二抗1∶100)37 ℃孵育1 h,TBS清洗3次后滴加DAPI封片,荧光显微镜拍片保存。
1.3 统计学方法
实验数据结果采用GraphPad Prism7软件分析,符合正态分布的定量数据结果以均数±标准差()表示。两组间比较采用t检验,多组间比较采用单因素方差分析,组间两两比较采用Dunnett-t检验,P<0.05为差异有统计学意义。
2 结果
2.1 shRNA载体的构建
为了成功敲降小鼠睾丸内Sox30 基因的表达,将特异性shRNA靶标序列分别设计在Sox30基因的1号外显子和5号外显子区域(图1A)。利用同源重组方法构建pSliencer-GFP-shSox30-1,pSliencer-GFP-shSox30-2 敲减载体和pSliencer-GFP-shScramble 对照载体,转化进原核细胞DH5α后PCR 筛选sense-Loop-antisense片段插入成功的阳性单克隆菌落,通用引物H1和CMV-R进行PCR扩增,目的条带为426 bp(图1B、C)。阳性菌落提取质粒进行测序,Sense-Loop-Antisense 序列测序结果正确的pSliencer-GFPshSox30-1和pSliencer-GFP-shSox30-2质粒用作后续敲降实验(图1D)。
2.2 shRNA敲降效率的验证
Sox30 是生殖细胞特异表达的转录因子,为了在体外验证两条shRNA 的敲降效率,将构建好的pSliencer-GFP-shScramble、pSliencer-GFP-shSox30-1或pSliencer-GFP-shSox30-2 重组质粒分别与Sox30过表达PRK5-HA-Sox30质粒共转入293T细胞中,未转染的细胞作为空白对照组。转染后继续培养36 h后收取细胞样品进行RT-qPCR 和Western blot 实验。重组质粒带有EGFP 标签,荧光显微镜下发现50%以上的293T细胞都是EGFP 阳性细胞(图2A),且实验组和对照组间的阳性细胞比例差异无统计学意义(图2B)。RT-qPCR 实验分别检测空白对照组、过表达组、对照组、实验组1、实验组2 中Sox30基因的mRNA 水平,结果显示两组实验组的Sox30转录本表达量显著下降,仅为过表达组的30%~40%(图2C)。Western blot结果显示两组实验组中Sox30蛋白表达量下降,约为过表达组的40%~50%(图2D)。其中实验组2在Sox30 mRNA水平和蛋白水平的敲降效率更高,用于后续睾丸组织病毒注射实验。
2.3 Sox30敲降小鼠模型的构建及验证
根据细胞实验中shSox30 的敲减效率结果,选取敲减效率较高的pSliencer-GFP-shSox30-2 重组质粒用于构建小鼠敲降模型。小鼠出生后18 d 睾丸生精管腔发育至圆形精子阶段,在第24天完成发育并逐步过渡至长型精子阶段。为了探究Sox30敲降对精子变形过程的影响,选取出生后24 d的野生型雄鼠,通过网微注射将pSliencer-GFP-shSox30-2 重组质粒注射至小鼠生精小管中,在重组质粒持续表达并敲降Sox30 转录本48 h 及7 d后,收集小鼠睾丸样品。本研究中提取小鼠睾丸总蛋白和RNA,分别进行RT-qPCR 和Western blot 实验,系统验证睾丸组织中Sox30 在mRNA 水平和蛋白水平的敲降效率。发现注射48 h后,实验组中Sox30 基因mRNA 水平下降,仅为对照组的20%,蛋白表达量约为对照组的一半,注射7 d后,实验组中Sox30 基因mRNA 水平为对照组的60%~70%,蛋白表达水平降低(图3A、B)。

图3 Sox30敲降小鼠睾丸mRNA和蛋白表达水平Figure 3 The expression of mRNA and protein level from mouse testis treated with shSox30 lentivirus
Sox蛋白家族分为A~H亚型。Sox30属于Sox H亚家族,且为唯一成员。为了明确shSox30 敲减系统是否存在脱靶效应,选取与Sox30 同源性最近的Sox D 亚家族成员Sox5、Sox6,分别进行RT-qPCR 和Western blot 实验。同对照组相比,注射48 h后,实验组中Sox5 和Sox6 的mRNA 水平无显著性差异,Sox5和SOX6的蛋白水平在两组中差异无统计学意义(图3C~E)。以上结果提示,本研究中的敲减体系不存在脱靶效应。
2.4 Sox30敲降小鼠生殖表型观察
选取注射后48 h 的对照组和实验组睾丸制备组织切片,根据EGFP 在生殖细胞中的荧光信号明确shRNA 感染的细胞类型。结果显示,注射48 h后,在前细线期精母细胞(preleptotene,PL)、偶线期精母细胞(zygotene,Zyg)、粗线期精母细胞(pachytene,Pac)、圆形精子细胞(round spermatid,RS)、减数第一次分裂中期细胞(metaphase I,MI)以及睾丸体细胞支持细胞(sertoli,Ser)中观察到绿色荧光信号(图4A)。以上提示本研究中的shRNA敲降系统成功感染多类型生殖细胞。进一步选取注射后7 d的对照组和实验组睾丸制备成组织切片,观察圆形精子发育进程。PNA可特异性识别圆形精子顶体蛋白,根据顶体蛋白覆盖面角度可区分圆形精子的发育时期。圆形精子发育至2~3 步时顶体蛋白染色呈1~2 个小点,发育至4~6步时顶体蛋白覆盖面角度逐渐延展至120°。免疫荧光结果显示,实验组中生精管腔内圆形精子发育阻滞在2~3步,无法正常发育至长型精子阶段(图4B)。苏木素-伊红染色结果显示,实验组睾丸生精小管Ⅶ~Ⅷ管腔中明显缺少长型精子(图4C)。
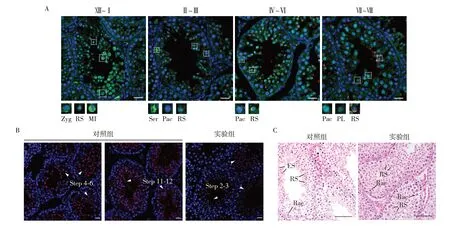
图4 Sox30敲降雄鼠生殖表型分析Figure 4 Analysis of the reproductive phenotype of male mice with Sox30 knockdown
3 讨论
本研究特异性设计靶向Sox30 基因转录本的shRNA 序列,将shRNA 构建进入pSilencer 载体中形成pSliencer-GFP-shSox30敲降载体。利用慢病毒包装系统形成侵染能力较强的慢病毒敲降载体,并通过网微注射将慢病毒敲降载体注射进出生24 d 雄鼠睾丸的输出小管,使其进入生精小管管腔。处理48 h和7 d后,分别收集小鼠睾丸进行实验验证。本研究利用RT-qPCR、Western blot 实验,从体外和体内水平验证shRNA体系具备高效敲减效率,且敲减效率至少可持续1 周。采用PNA 进行免疫荧光,结合睾丸组织免疫组化实验,证实了该体系成功敲降生殖细胞重要转录因子Sox30 的表达,观察到圆形精子发育阻滞在第2~3步,Ⅶ~Ⅷ期生精管腔中无长型精子,实验组中产生了与Sox30 全敲雄性小鼠类似的生殖表型。
精子发生是一个连续且有序的生理过程,涉及精原干细胞的自我更新、精母细胞的减数分裂,以及单倍体精子的变形过程[18-19]。某一精子发生相关基因可能在精子发生的多个阶段都发挥功能。传统的基因敲除模型可能在生精细胞发育分化的较早阶段就敲除了目的基因而导致精子发生阻滞在早期阶段,导致无法研究这一基因在精子发生晚期阶段的表型和功能。借助本平台可以根据实验需要,在第一波生精波发生的相应时间点将慢病毒敲降载体注射进入小鼠睾丸生精管腔中,从而可以达到在特定时间点完成目的基因的敲降工作。传统的基因敲除模型通过CRISPR/Cas9等基因编辑技术进行基因打靶工作,但是此技术存在脱靶效应。同时,基因编辑技术是在基因组层面上进行操作,基因打靶脱靶至基因组其他基因位点后可能导致不可逆的功能和表型改变。而且,脱靶后的位点很难被检测到,可能会产生实验者无法解释的实验结果。慢病毒敲降平台在基因敲降应用上具有显著优势。一方面,该平台靶向转录组mRNA 设计shRNA 敲降序列,即使出现脱靶效应也不会改变小鼠的基因组序列,产生可遗传的变异;另一方面,慢病毒敲降载体注射至小鼠睾丸生精小管中,仅在注射部位局部发挥敲降功能,并不会通过血睾屏障传递至全身,脱靶效应也只发生在睾丸中。虽然直接注射siRNA或者反义寡核苷酸也可以达到敲减基因表达的功能,但是有研究指出siRNA 在注射后48 h 达到最高敲降效率,此后由于无法持续表达扩增而慢慢失效。而本研究注射的是shRNA载体,在侵染细胞后会迅速入核并在细胞核内持续表达加工形成前体shRNA,再转位至胞质经过加工获得siRNA,敲降持续时间较久。
本研究前期已构建Sox30 基因全身性敲除小鼠,并系统阐述了Sox30全敲小鼠的睾丸表型,以及Sox30 在精子发生过程中的作用机制。在Sox30 全敲小鼠睾丸中,精子发生阻滞在2~3 步的圆形精子阶段。然而Sox30蛋白的表达一直持续到精子变形的晚期,Sox30全敲小鼠模型无法研究其在精子变形的中晚期是否发挥作用。因此,利用慢病毒敲降平台构建的Sox30敲降小鼠模型为研究Sox30在精子变形晚期的功能机制奠定了研究基础。
猜你喜欢
中国临床医学影像杂志(2022年5期)2022-07-26
右江医学(2021年9期)2021-10-22
昆明医科大学学报(2021年1期)2021-02-07
中国生殖健康(2019年7期)2019-01-06
中国生殖健康(2019年6期)2019-01-06
癌变·畸变·突变(2016年5期)2016-08-22
中国男科学杂志(2016年9期)2016-03-20
安徽医药(2014年9期)2014-03-20
遗传(2014年3期)2014-02-28
生殖医学杂志(2013年3期)2013-03-11
