
热修饰大豆分离蛋白与改性魔芋胶糖基化对产物抗水化特性的影响研究
2023-06-15 07:25:02郑雅丹游寅寅杨鹏顾继鹏冯魏陈玉峰刘书来丁玉庭
包装工程 2023年11期
郑雅丹,游寅寅,杨鹏,顾继鹏,冯魏,陈玉峰,刘书来,丁玉庭
(1.浙江衡美健康科技股份有限公司,杭州 311100;2.浙江工业大学 食品科学与工程学院,杭州 310014)
随着生活水平的提高,饮食结构及生活方式的改变导致超重和糖尿病患者日益增多。高蛋白高膳食纤维冲调食品因其具有富营养、低热量、食用方便等特点备受关注[1]。都阳等[2]通过不同粉碎方式的高膳食纤维组合制备了营养丰富流动性好的代餐粉;甘聃[3]通过胶体和油脂添加种类及用量优化制备了分散性好、口感顺滑的代餐粉;刘先娥[4]以抗性淀粉为基料制备了感官特性优良的复合膳食纤维代餐粉。然而,基于高蛋白和高膳食纤维改性的代餐冲调食品研究鲜有报道。
大豆分离蛋白(SPI)和魔芋胶(KGM)均是制备高蛋白高膳食纤维复合物食品的优选原料[5-6]。然而,较高浓度SPI 易产生絮凝或形成凝胶网络结构[7],当冲调类产品中的SPI 与KGM 混合冲调后,因水化作用会形成较为致密的蛋白纤维复合网络结构,易出现黏度高和流动性差等问题,无法满足吞食类饮品的良好适口性能要求。Wang 等[7]通过将低浓度大豆蛋白(1.0%)预热,使大豆蛋白在热修饰后的抗水化抗聚集能力得到提高。罗清楠等[8]运用酸降解方法,以特性黏度为表征,对KGM 进行酸降解,经过处理后的KGM 随着分子质量降低其黏度也随之降低。糖基化修饰可改善天然蛋白质的功能特性[9]。谷氨酰胺转氨酶(TG 酶)可使蛋白质肽链相应基团发生交联,同时促进糖分子与蛋白质发生共价结合,形成糖基化复合物[10]。本研究通过改性SPI 和可溶性膳食纤维KGM,并进行糖基化和TG 酶耦合交联反应,制备具有黏度低和良好吞食性能的高蛋白高膳食纤维冲调型食材,以期为奶昔等功能多元化代餐食品的开发提供一定指导。
1 实验
1.1 材料与试剂
主要材料与试剂:SPI,河南万邦实业有限公司;KGM(KT12 型),湖北强森魔芋科技有限公司;柠檬酸,浙江一诺生物科技有限公司;氢氧化钙,浙江一诺生物科技有限公司;液体型谷氨酰胺转氨酶(120 U/g),上海青瑞食品科技有限公司;胃蛋白酶(3 000 U/g)、胰蛋白酶(400 U/g),上海阿拉丁生物科技股份有限公司;胶态微晶纤维素,河北食化食说原料有限公司。
1.2 仪器与设备
主要仪器与设备:AR2130 电子精密天平,上海里衡仪器仪表有限公司;HYJD 超纯水器,杭州永洁达净化科技有限公司;HH–1 数显恒温水浴锅,江苏省金坛市江南仪器厂;PHS–3C pH 计,上海精密科学仪器有限公司;HR2860 打浆机,德国飞利浦有限公司;SCIENTZ–12N 超低温冷冻干燥机,宁波新芝生物科技有限公司;FA25 高速分散机,上海Fluko流体机械制造有限公司;RE52–99 旋转蒸发器,上海亚荣生化仪器厂;YC–1800 实验室低温喷雾干燥机,上海雅程仪器设备有限公司;NDJ–5S 旋转黏度计,上海平轩科学仪器有限公司;MCR302 旋转流变仪,奥地利安东帕有限公司;UV–VIS 紫外可见分光光度计,上海美谱达仪器有限公司;CR21GⅡ高速离心机,日立Hitachi 公司。
1.3 制备方法
1.3.1 热处理大豆分离蛋白的制备
参照Wang 等[7]的实验方法稍做修改。将质量分数为1%的SPI 溶于水并搅拌30 min 使其混合均匀,于90 ℃搅拌60 min 后冷却至室温,加入柠檬酸调节体系pH 至4.5 后抽滤收集沉淀,加入饱和澄清石灰水将体系pH 调整为7.0 后真空冷冻干燥72 h,过筛得到热处理大豆分离蛋白(TS)。
1.3.2 预处理KGM 的制备
1.3.2.1 酸降解KGM 制备
参照Makabe 等[11]方法制备低分子质量KGM。将质量分数为10%的KGM 缓慢加入柠檬酸溶液中。其中柠檬酸的浓度为每克KGM 添加0.16 mmol,搅拌30 min 使其混合均匀,80 ℃加热搅拌3 h 使其酸解。接着向体系中倒入2 倍体积的体积分数为95%的乙醇,打浆搅拌均匀,静置沉淀后过滤收集沉淀,重复上述操作并合并沉淀物。最后真空冷冻干燥72 h,过筛得到酸降解KGM(HK)。
1.3.2.2 碱脱乙酰基KGM 的制备
参照Li 等[12]的方法加以改进。精确称取KGM分散在2 倍体积的去离子水中,在室温下搅拌并溶胀30 min。立即加入饱和澄清石灰水,其中氢氧化钙的浓度为每克KGM 添加0.03 mmol,使KGM 在体系中的质量分数为10%,搅拌10 min 使其混合均匀,95℃水浴反应1.0 h。待脱乙酰基反应完成后,用一系列浓度梯度乙醇溶液(体积分数为50%、75%、95%)进行样品脱水。所得产物冷冻干燥72 h,过筛得到碱脱乙酰基KGM(OHK)。
1.3.3 酶交联耦合糖基化制备SPI-KGM 复合物
质量分数为4%的TS 溶于水,分别加入质量分数为1%的KGM、HK 或OHK,再加入质量分数为0.15%为微晶纤维素作为抗结剂及质量分数为0.06%的谷氨酰胺转氨酶(TG 酶)以促进蛋白交联,混合物以10 000 r/min 高速剪切分散1 min。在40 ℃下旋转蒸发1 h 使底物发生一定糖基化反应,并将体系固形物浓缩至(8.0±0.2)%,调节体系pH 至5.5,喷雾干燥后得到大豆分离蛋白–魔芋胶复合物(TS–K、TS–HK 和TS–OHK)。
1.3.4 接枝度测定
按照Li 等[13]方法进行测定。加入磷酸缓冲溶液将样品蛋白质量浓度调节至2.0 mg/mL。取4.0 mL 邻苯二甲醛试剂于试管中,加入200 μL 样品,涡旋振荡使其混合均匀,置于35 ℃水浴锅中反应2 min,于340 nm处测定吸光值。以赖氨酸代替样品,以相同的方法做标准曲线。接枝度(DG)以式(1)进行计算。
式中,C0为糖基化修饰前体系游离氨基的含量,mol/L;C1为糖基化修饰后体系游离氨基的含量,mol/L。
1.3.5 傅里叶红外光谱测定
用傅里叶红外光谱对SPI–KGM 复合物粉末状态中蛋白的一、二级结构进行分析。称取少量干燥样品粉末与溴化钾粉末充分混合、研磨压片后于红外光谱仪舱室中进行全波长扫描。波数范围为400~4000 cm−1,分辨率为4 cm−1,扫描次数为32 次。采用Omnic 8.0和Peakfit 4.12 软件进行分析[14]。
1.3.6 表面疏水性与接触角测定
参照Li 等[15]的方法并稍加改进,取0.2 g 样品溶于20 mL 去离子水中,离心取4 mL 上清液,加入80 μL质量浓度为1 mg/mL 的溴酚蓝溶液并混合均匀,室温反应10 min 后,离心取上清液稀释5 倍,在595 nm测定样品吸光度。以未加样品的溴酚蓝溶液作为空白样。表面疏水性采用式(2)进行计算:
式中:H0为表面疏水性,μg;A0为对照组的吸光度;A1为实验组的吸光度。
参考支雅雯等[16]的方法,使用接触角测定仪测定接触角,将大豆分离蛋白–魔芋胶复合物粉末压片后,置于测定仪上,测量范围为0~180°,滴水量为0.5 μL。
1.3.7 溶解度测定
参考夏轩泽等[17]方法稍作改动。配制100 mg/mL的大豆分离蛋白–魔芋胶复合物溶液,以10 000 r/min的转速高速分散1 min 后,取10 mL 溶液进行离心(3 500 r/min、15 min、25 ℃)。分别检测离心前后分散体系中的蛋白含量。蛋白含量均采用双缩脲法进行测定,并以牛血清蛋白作为标准蛋白。溶解度采用式(3)表示。
式中:m0为离心前样品蛋白含量,mg/mL;m1为离心后上清液蛋白质含量,mg/mL。
1.3.8 表观黏度测定
分别用25 ℃和100 ℃去离子水以固液比10 ∶1将SPI–KGM 复合物进行冲调溶解,冷却后使用数字黏度计测量复水后的黏度。测量期间所有样品的温度均保持在25 ℃,选用3 号转子,转速控制在12 r/min。
1.3.9 消化特性表征
参考Silva 等[18]实验方法并稍作改动,分别制备不同离子浓度胃消化液(Stimulated Gastric Fluid,SGF)和肠道消化液(Simulated Intestinal Fluid, SIF)备用。
胃消化阶段:取20 mL 质量分数为6%的复合物分散液,加入至20 mL SGF 中,并加入3 mL 胃蛋白酶液(50 mg/mL),用浓度为1 mol/L 盐酸溶液调节pH 至1.5,在37 ℃恒温水浴震荡(转速为150 r/min)2 h。
肠消化阶段:取20 mL 胃消化后分散液,分别加入20 mL SIF,2 mL 浓度为160 mmol/L 胆盐和3 mL胰蛋白酶液(100 mg/mL),用浓度为1 mol/L 氢氧化钠调节pH 至7.4,并用浓度为0.05 mol/L 的氢氧化钠溶液滴定以维持体系pH 恒定,在37 ℃恒温水浴震荡(转速为150 r/min)2 h。
1.3.9.1 消化溶胀率测定
参考王文霞等[19]通过比较消化前后质量比测量溶胀率。分别称取等量的SPI–KGM 复合物,记为m0;分散于去离子水中,静置溶胀30 min,过滤并称量得到溶胀后质量;将复合物干粉溶入消化模拟液中进行体外消化,消化后灭酶处理的样品过滤称量,得到消化后质量。溶胀率计算式如式(4)所示。
式中:m0为样品干粉的质量,g;m1为样品溶胀或消化后的质量,g。
1.3.9.2 消化黏度测定
消化后的混合物按照1.3.8 节方法进行黏度测定,仅进行25 ℃下的测量。
2 结果与分析
2.1 魔芋胶改性方式对SPI–KGM 复合物接枝度的影响
KGM 与SPI 发生反应形成的SPI–KGM 复合物的接枝度受魔芋胶改性方式的影响。由图1 可知,KGM 的接枝度从0%(TS)逐渐上升至(39.71±1.11)%(TS–K),表明 KGM 与 SPI 发生反应,形成了SPI–KGM 复合物。同样,KGM 经酸碱改性处理后,TS–HK 和TS–OHK 组接枝度分别提高至(48.84±1.06)%和(44.50±0.74)%。其中酸降解改性KGM使得分子质量变小,具有更多还原性羰基,更易与蛋白分子发生糖基化反应[20];碱改性处理发生脱乙酰基反应,导致KGM 空间位阻效应降低,有利于嵌入蛋白质内部,促进了糖基化反应。

图1 魔芋胶改性方式对SPI–KGM 复合物接枝度的影响Fig.1 Effect of KGM modification method on the grafting degree of SPI-KGM composite
2.2 SPI–KGM 复合物傅里叶红外光谱解析
傅里叶红外光谱可用于研究SPI 构象变化和糖基化反应。如图2 所示,不同KGM 改性方式的SPI–KGM 复合物红外光谱图与对照组SPI 与TS 相比,糖基化后的复合物在1 500~1 350 cm−1间的吸收会增强(如表1 所示),这主要是由于C−N 伸缩和N−H 变化产生的。此外,在1 260~1 000 cm−1处有明显的吸收峰,这是因为KGM 中C−O−C 糖苷键的伸缩振动,也显示出SPI 与KGM 发生了糖基化反应[21]。

表1 不同SPI–KGM 复合物红外峰强度统计Tab 1 Statistics of infrared peak intensity of different SPI-KGM composites

图2 不同魔芋胶改性方式的SPI–KGM复合物红外光谱图Fig.2 FT-IR spectrum of SPI-KGM compositesbydifferent KGM modification methods
将红外光谱中酰胺Ⅰ带进行去卷积和多峰拟合来计算SPI–KGM 复合物二级结构,如表2 所示。TS相较于天然SPI 的α–螺旋从33.06%下降为31.64%,β–折叠从19.85%显著增大至30.73%,说明热处理使TS 蛋白质发生一定程度的变性聚集。与对照TS 的无规则卷曲17.28%相比,加入KGM 后,TS–K、TS–HK和 TS–OHK 的无规则卷曲从 23.19%分别增加至28.24%、26.21%和29.08%,含量明显增大。说明,随着糖基化反应过程中多糖与蛋白质的羰基和ε–氨基之间的缩合,分子内氢键被破坏造成α–螺旋结构进一步降低,从而导致α–螺旋结构伸展成β–折叠、β–转角或无规则卷曲等二级结构[13]。

表2 不同魔芋胶改性方式的SPI-KGM 复合物二级结构相对含量比较Tab 2 Comparison of the relative content of the secondary structure in SPI-KGM compositesby different KGM modification methods %
2.3 SPI–KGM 复合物表面疏水性及接触角的比对分析
不同魔芋胶改性方式对SPI–KGM 复合物表面疏水性及接触角产生一定影响。如图3 所示,天然SPI的表面疏水性为(25.26±1.81)μg,引入KGM 反应后,TS–K、TS–HK 和TS–OHK 的表面疏水性分别增加至(37.16±1.18)、(34.87±0.58)和(48.75±1.16)μg。这可能是基于SPI 与KGM 反应生成的复合物的空间结构发生变化,使隐藏在分子内部的疏水性基团暴露所致[22]。

图3 魔芋胶改性方式对SPI–KGM 复合物表面疏水性的影响Fig.3 Effect of KGMmodification methodon the hydrophobicity of SPI–KGMcomposite surface
接触角θ代表亲水性和疏水性的大小,接触角越大显示疏水性越强[23]。由图4 可知,天然SPI 接触角为(58.88±0.54)°,显示疏水性较弱,经过TG 酶交联耦合糖基化反应后,TS–K、TS–HK 和TS–OHK 接触角分别增大至(65.52±1.66)°、(62.59±0.58)°和(68.47±1.29)°,表明疏水性得到明显增强,这与上述表面疏水性结果一致。由此可知,大豆分离蛋白–魔芋胶复合物的抗水化性有了一定程度的提高。

图4 魔芋胶改性方式对SPI–KGM 复合物接触角的影响Fig.4 Effect of KGMmodification methodon the contact angle of SPI–KGMcomposite
2.4 魔芋胶改性方式对SPI–KGM 复合物溶解特性的影响
溶解特性一定程度反映了复合物在食用时的冲调性能。如图5 所示,与天然SPI 溶解度(68.22±1.39)%相比,KGM 经过酸降解与碱改性之后,SPI–KGM 复合物的溶解度从(76.75±1.55)%(TS–K)增加到(85.71±0.70)%(TS–HK)和(86.98±2.30)%(TS–OHK)。由此可知,经酶交联耦合糖基化修饰改性后,特别是随着糖基化修饰程度增强,多羟基多糖与蛋白质接枝有利于复合物溶解特性的增加。这一定程度反映了复合物的冲调性能可满足各类食品的应用场景。

图5 魔芋胶改性方式对SPI–KGM 复合物溶解度的影响Fig.5 Effect of KGM modification method on solubility of SPI-KGM composite
2.5 魔芋胶改性方式对SPI–KGM 复合物表观黏度的影响
黏度是液体食品的重要评价指标,蛋白发生水化作用后颗粒聚集会使得体系变黏稠。由图6可知,SPI与KGM直接混合溶解后的黏度为(2 616.83±66.71)mPa·s,而用100 ℃的热水冲调后,其黏度则达到(3607.50±56.29)mPa·s(S–K),导致吞食适口性较差;这主要是由于高浓度下SPI 分子易发生水化而展开并聚集,从而形成高度有序的蛋白凝胶三维网络结构[24]。与TS–K 相比,天然SPI 经过热诱导和酶交联耦合糖基化修饰后,TS–HK 常温和 100 ℃冲调后的体系黏度分别降低至(453.67±15.33)mPa·s 和(420.33±9.50)mPa·s。同样,碱改性KGM 脱乙酰基后与TS 作用得到的复合物,TS–OHK 常温和100 ℃冲调后的体系黏度分别降低至(577.67±58.64)mPa·s 和(461.33±16.29)mPa·s。实验表明,经热修饰后的SPI 与改性KGM 生成的糖基化复合物具有较好的抗水化能力和较低的体系黏度,这对开发低黏度适口性好的冲调食品具有广阔的应用前景。
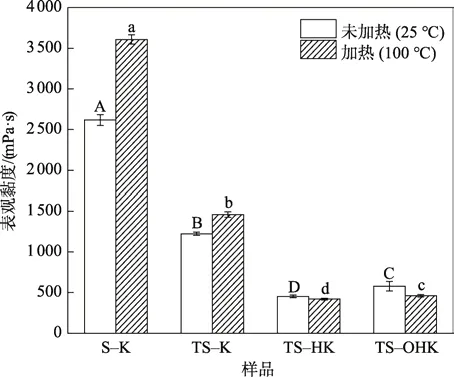
图6 冲调温度对SPI–KGM 复合物分散体系表观黏度的影响Fig.6 Effect of reconstitution temperature on apparent viscosity of SPI-KGM compositedispersion system
2.6 魔芋胶改性方式对SPI–KGM 复合物消化溶胀特性的影响
食物的溶胀和黏度是影响饱腹感的重要因素,通过影响胃扩张和胃排空来提升饱腹感[25–30]。因此,在体外消化模拟实验中选用溶胀率与黏度指标评价SPI–KGM 复合物的饱腹感。复合物冲调后较低的溶胀率将有利于等量水分下溶质比的提高和吞食时低黏适口性的需求。如图7 所示,与SPI 和KGM 直接混合组的溶胀率(26.57±0.82)%(S–K)相比,经酶交联耦合糖基化修饰后的大豆分离蛋白–魔芋胶复合物溶胀率显著降低(P<0.05)。其中TS–OHK 组溶胀率最小,为(16.21±1.61)%,其次是TS–HK 组,溶胀率为(16.52±0.62)%。

图7 SPI–KGM 复合物的溶胀率随消化时间变化的动力学曲线Fig.7 Kinetic curve of swelling rate of SPI-KGM composite with digestion time
在pH 值为1.5的胃模拟体系中消化后,与对照S–K组的溶胀率(24.71±1.31)%相比,SPI–KGM 的复合物TS–K、TS–HK 和TS–OHK 的溶胀率均有一定程度的提高,分别为(26.73±1.56)%、(21.24±0.96)%及(16.86±1.05)%。这是基于胃中蛋白酶水解包覆复合物的蛋白质,使复合物中魔芋多糖暴露导致的。随着消化时间延长,对照S–K 组的溶胀率出现逐渐降低的趋势,而经糖基化后的复合物均表现出了一定的溶胀率上升趋势。说明复合物具有消化持续溶胀的能力,特别是在消化4.0 h 后,TS–OHK 组仍然保持稳定上升的趋势。
2.7 复合物的黏度消化动力学变化曲线
消化黏度反映食物在消化体系中的黏滞性和胃部排空情况,一定程度影响着餐后饱腹感。如图8 可见,SPI 与KGM 直接混合后黏度较大,起始值为(1802.17±73.24)mPa·s(S–K),经模拟体系4 h 后,降至(1048.17±17.47)mPa·s,消化体系的黏度变化较大,而经糖基化后的复合物整体消化黏度变化均较小。综上消化溶胀率和消化黏度的变化可知,热修饰SPI 与改性KGM 糖基化复合物作为冲调食材可维持持续的饱腹感优势。

图8 SPI–KGM 复合物的黏度随消化时间变化的动力学曲线Fig.8 Kinetic curve of viscosity of SPI-KGM composite with digestion time
3 结语
本研究利用热处理SPI 在TG 酶促进下发生交联反应同时耦合糖基化,将酸碱处理后KGM 包裹在三维网络结构内部,制备出具有一定抗水化特性的SPI–KGM 复合物。结果表明,制备的高蛋白–高膳食纤维复合物具有较好的抗水化和冲调低黏度特性。在模拟消化过程中,复合物表现出稳定的消化溶胀与黏度特性,可很地好满足吞食低黏度和胃中强饱腹感的代餐需求,其中经碱改性后KGM 反应复合物作为优先组别。因此,热修饰大豆分离蛋白与改性魔芋胶糖基化复合物作为一种健康代餐食品原料具有良好的应用前景。
猜你喜欢
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
今日农业(2019年14期)2019-09-18 01:21:42
中成药(2018年7期)2018-08-04 06:04:18
中成药(2018年3期)2018-05-07 13:34:18
金色少年(奇趣科普)(2016年9期)2016-10-21 07:26:49
中国酿造(2016年12期)2016-03-01 03:08:27
医学研究杂志(2015年12期)2015-06-10 06:57:46
中国粮油学报(2014年7期)2014-02-06 01:33:01
现代检验医学杂志(2014年1期)2014-02-06 01:29:26
现代检验医学杂志(2014年2期)2014-02-02 02:40:48
