
4,4′-偶氮-1,2,4三唑与4,4′-联-1,2,4-三唑含能性质的DFT研究
2023-06-13 10:17:50章佩佩何晓卉程龙玖
火炸药学报 2023年5期
黄 鑫,章佩佩,卜 舒,何晓卉,王 昆,程龙玖
(安徽大学 化学化工学院化学系,安徽 合肥 230601)
引 言
作为一种新兴的高能材料,含能金属有机框架(EMOFs)因其高密度、良好的热稳定性和高爆热而成为研究热点[1-3],在烟花、起爆药、炸药和高能复合材料等领域具有广阔的应用前景[4-7]。近年来,以4,4′-偶氮-1,2,4-三唑(ATRZ)以及4,4′-联-1,2,4-三唑(BTRZ)为能量配体的含能金属有机框架(EMOFs)被大量的合成和研究[8-9]。
上述大量配体-配位环境的构效关系研究均是基于三唑环形成的两种不同的含能配体形成的配合物。对于ATRZ和BTRZ作为含能配体,其优异的含能特性、配位性能与其分子结构和晶体结构之间的关系有待于进一步研究。本研究基于密度泛函理论,对分子体系和周期性体系的ATRZ和BTRZ进行分析计算,并使用从头算分子动力学方法对两种配体结构进行分解轨迹的研究,为其在金属配合物中构效关系的研究提供依据。
1 量子化学方法
1.1 方法概述
本研究对于分子体系的4,4′-偶氮-1,2,4-三唑(ATRZ)和4,4′-联-1,2,4-三唑(BTRZ),均在Gaussian 16软件包[16]中用TPSS/6-311++G(3d,2p)方法进行了结构优化与性质的计算。根据理论生成焓值与实验值的比较确定了该DFT方法。
所有的晶体结构均使用Materials Studio软件中的CASTEP程序[17]计算,利用GGA-PBE的广义梯度近似,对ATRZ晶体和BTRZ晶体进行了优化。使用PBE型的超软赝势来描述价电子,具体为C(2s22p2)、N(2s22p3)以及H(1s1)。ATRZ和BTRZ的布里渊区[18]分别通过5×4×3和3×3×2格点进行描述。在优化过程中,两种化合物的波函数截止能量均设定为570 eV。将BFGS算法应用于原子位置的结构弛豫[19],直到原子剩余力和应力分别小于0.03 eV/Å和0.05 GPa。基于优化结构,晶体的能带与态密度通过HSE06方法进行了研究。

2 结果与讨论
2.1 ATRZ和BTRZ分子体系
2.1.1 几何结构与电荷分布
ATRZ和BTRZ的分子体系通过TPSS/6-311++G(3d,2p)水平优化,如图1所示。以优化结构为基础,部分化学键的键长、Wiberg键级和ADCH电荷总结于表1。相比于4H-1,2,4-三唑,ATRZ和BTRZ环内C—N与N—N键的键长趋于平均化,说明存在共轭作用;偶氮N=N键长(1.259 Å)处于1.25~1.27 Å之间,从ESP图可以发现ATRZ中偶氮键的引入使得N3、N8、N9、N10间的电荷平均化,证明了ATRZ中更强的共轭作用。

表1 ATRZ与BTRZ的部分化学键的键长、Wiberg键级与ADCH电荷
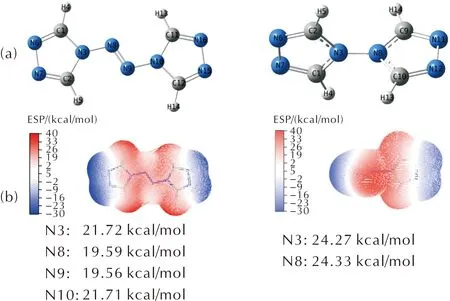
图1 ATRZ(左)和BTRZ(右)的(a)结构;(b)静电势(ESP)
对于ATRZ和BTRZ,在相同理论水平下使用Multiwfn程序[22]计算原子偶极矩校正的Hirshfeld布居电荷(ADCH)[23]。如表1所示,ATRZ三唑环上N3原子的ADCH电荷(-0.226 a.u)值最小,说明其反应活性最高,更容易与金属形成配合物,而BTRZ上的N3、N4原子的电荷(-0.228 a.u)相同,说明二者反应活性相同且大于ATRZ上的N3。所有碳原子和氢原子上均为正电荷。
根据Hirshfeld表面和2D指纹图中可以确定原子间紧密接触的方向、强度和距离,见图2。如图2所示,Hirshfeld表面的红色斑点代表形成氢键的位点,ATRZ的红点在同一平面上,有利于降低撞击敏感度,而BTRZ的红点几乎垂直[24]。指纹图的原子间紧密接触中,二者都是N—H键强度最高且BTRZ的N—H峰更加尖锐,说明BTRZ的N原子作为氢键供体的能力更强,化学反应活性更强,与文献记录相符[9]。

图2 ATRZ和BTRZ的Hirshfeld表面与二维指纹图
2.1.2 生成焓
等键反应方法[25]已成功用于计算生成焓,为求得ATRZ与BTRZ的生成焓,构建等键反应式,如图3所示。

图3 ATRZ与BTRZ的等键反应式
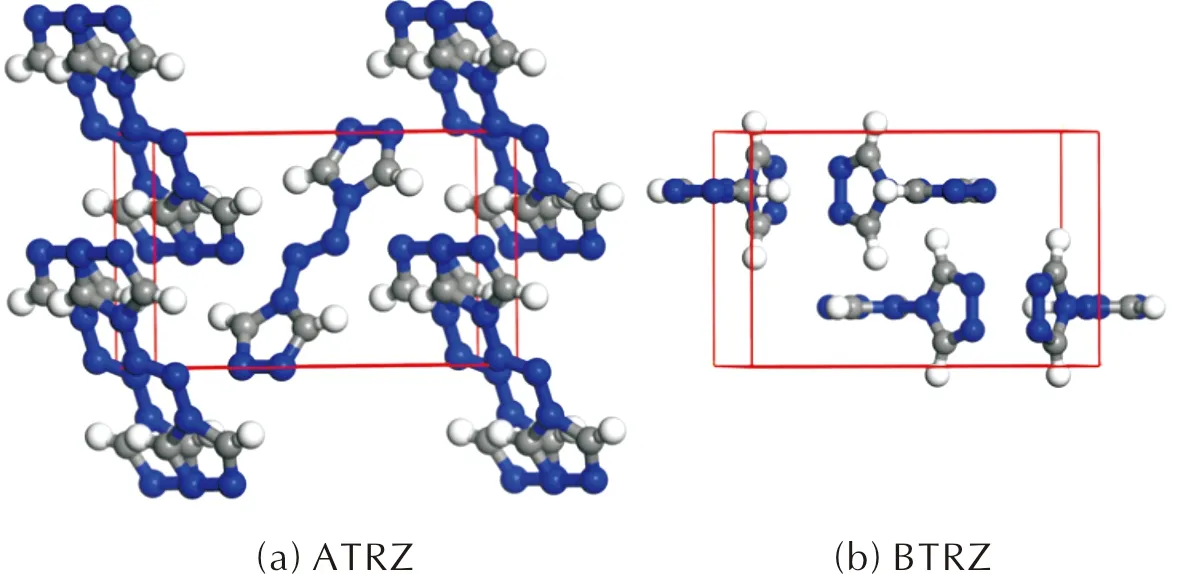
图4 ATRZ晶体和BTRZ晶体的优化结构(灰色:C;蓝色:N;白色:H)
ΔrH298 K=∑ΔfHP-∑ΔfHR
(1)
ΔrH298 K=ΔE0+ΔZPE+ΔHT+ΔnRT
(2)
NH3的实验生成焓(-45.90 kJ/mol)及N2H4的实验生成焓(95.40 kJ/mol)可由NIST数据库[26]或文献中查阅得到,E0、ZPE、HT可通过DFT计算方法得到,联立公式(1)、(2)即可得出等键反应的反应焓和目标产物的生成焓。对于实验生成焓未知的化合物,需要使用原子化能方法[27],利用已知的孤立原子的生成热,通过计算原子化反应能来预测分子的生成焓。分子AxByCz的原子化反应式如下:

ΔrH298 K=xΔfH298 K(A)+yΔfH298 K(B)+
zΔfH298 K(C)-ΔfH298 K(AxByCz)
(3)
ΔrH298 K=ΔE0+ΔHT
(4)
式中:ΔfH298 K(A)、ΔfH298 K(B)、ΔfH298 K(C)与ΔfH298 K(AxByCz)分别是原子A、B、C与分子AxByCz在298 K下的生成焓。
从ATRZ和BTRZ的分子结构(图1)可以发现,二者都含有两个4H-1,2,4-三唑环,至今尚未有文献报道过4H-1,2,4-三唑环生成焓的实验值,而其同分异构体1H-1,2,4-三唑可在NIST数据库中查询到实验值(193.70 kJ/mol),故可将这一实验值作为基准,比较部分泛函方法和基组的适用性。表2为计算水平下1H-1,2,4-三唑生成热的理论预测值及与实验值的偏差。
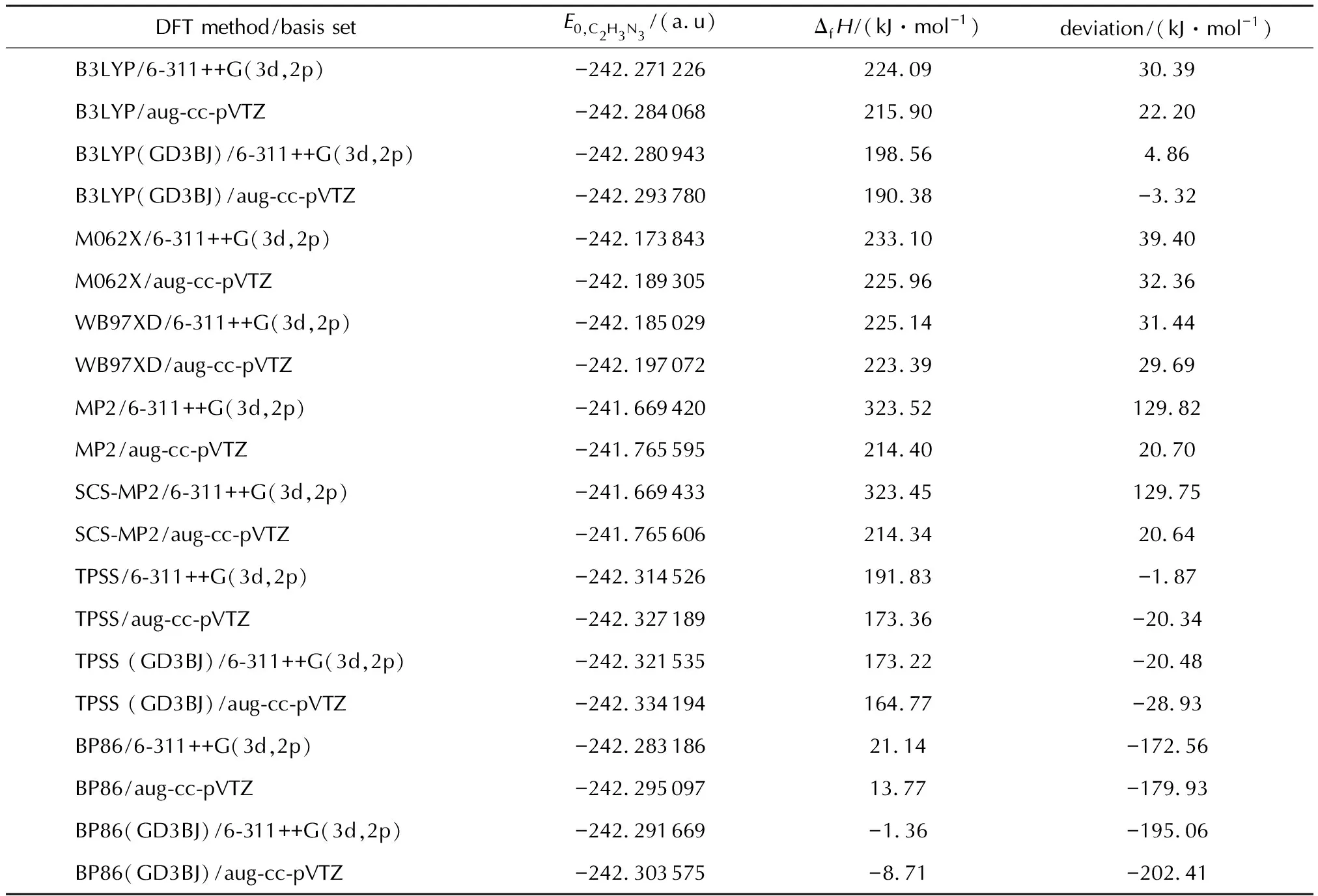
表2 在各计算水平下1H-1,2,4-三唑的生成热的理论预测值及与实验值的偏差
从表2中可以看出,相比于杂化泛函B3LYP、M062X与WB97XD,使用高斯基组和赝势基组皆与实验值符合性较好,说明其对基组的依赖性较小,基组影响小,重复性好,而且对B3LYP泛函加上GD3BJ校正[28-31]后,与实验值更加吻合。纯泛函BP86使用这两种基组和GD3BJ校正的结果准确性差,对于Post-Hartree-Fock泛函(MP2、SCS-MP2),选择哪种基组严重影响结果的准确性,在使用这种方法时,均需要严格测试基组。根据表2,在TPSS/6-311++G(3d,2p)水平下计算得出1H-1,2,4-三唑的生成焓(191.83 kJ/mol)最接近于实验值,偏差最小。除了对1H-1,2,4-三唑进行了泛函方法和基组的测试,本研究对1-H四唑(实验值为321.1 kJ/mol)和1-氨基四唑(实验值为323.8 kJ/mol)也分别进行了类似的测试,结果表明TPSS/6-311++(3d,2p)方法得到的计算值与实验值最为接近,偏差分别为-0.9和-2.5 kJ/mol。
因此,基于TPSS/6-311++(3d,2p)方法,可得4H-1,2,4-三唑生成焓为215.91 kJ/mol,N4H4的生成焓为283.64 kJ/mol,从而计算出ATRZ的生成焓为836 kJ/mol,BTRZ的生成焓为608 kJ/mol。显然,ATRZ比BTRZ的生成焓多228 kJ/mol,是因为ATRZ中偶氮键的引入使得三唑环之间的共轭程度增加,且ATRZ的含氮量(68.3%)大于BTRZ(61.8%),大大提高了ATRZ的生成焓。
2.1.3 密度、爆轰参数
含能材料的密度与爆轰性能参数密切相关。本研究采用实验所得的晶体密度值估算了ATRZ和BTRZ的爆速(D)及爆压(P)。对于仅含C、H、O、N元素的含能材料通常在计算出理论密度和生成焓的基础上,使用Kamlet-Jacobos方程(5)~(6)求得爆速和爆压[32-34]:
D=1.01(NM1/2Q1/2)1/2(1+1.30ρ)
(5)
P=1.558ρ2NM1/2Q1/2
(6)
式中:N为每克炸药爆轰生成气体产物的量(mol/g);M为爆炸气体产物的摩尔质量(g/mol);Q为每克炸药的最大爆热(J/g);ρ为装药密度(g/cm3)。
基于实验所得的晶体密度[35-36],将已得到的生成焓与密度的数值代入Kamlet-Jacobos方程即可得出相应的爆轰参数理论值[37],如表3所示,理论结果与部分实验结果的比较说明本次计算是可靠的。尽管二者均为高氮含能材料的结构基元,但表3的结果表明,ATRZ的爆热、爆速及爆压均优于BTRZ。
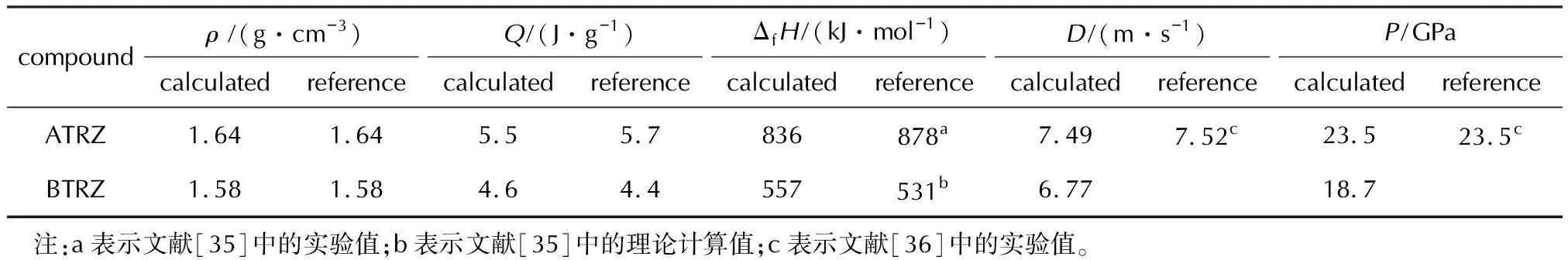
表3 TPSS/6-311++G(3d,2p)水平下ATRZ与BTRZ的生成焓、密度与爆轰参数
2.2 晶体结构与性质
基于实验结构[8-9],在Materials Studio的CASTEP模块中使用GGA-PBE方法优化了ATRZ和BTRZ的晶体结构,结果如图5所示,晶胞参数、典型键长和键角在表4中列出,并与实验结果进行了比较。ATRZ采用单斜晶系P21/N空间群,单个晶胞中有2个ATRZ分子。每个ATRZ分子中的两个三唑环在同一个平面上,其二面角为180°。对于BTRZ,单个晶胞中有4个BTRZ分子,并且每个BTRZ分子中的两个三唑环几乎垂直,二面角为91.96°。

表4 ATRZ和BTRZ晶体的晶系、点群、能带、晶胞参数、部分键长和键角
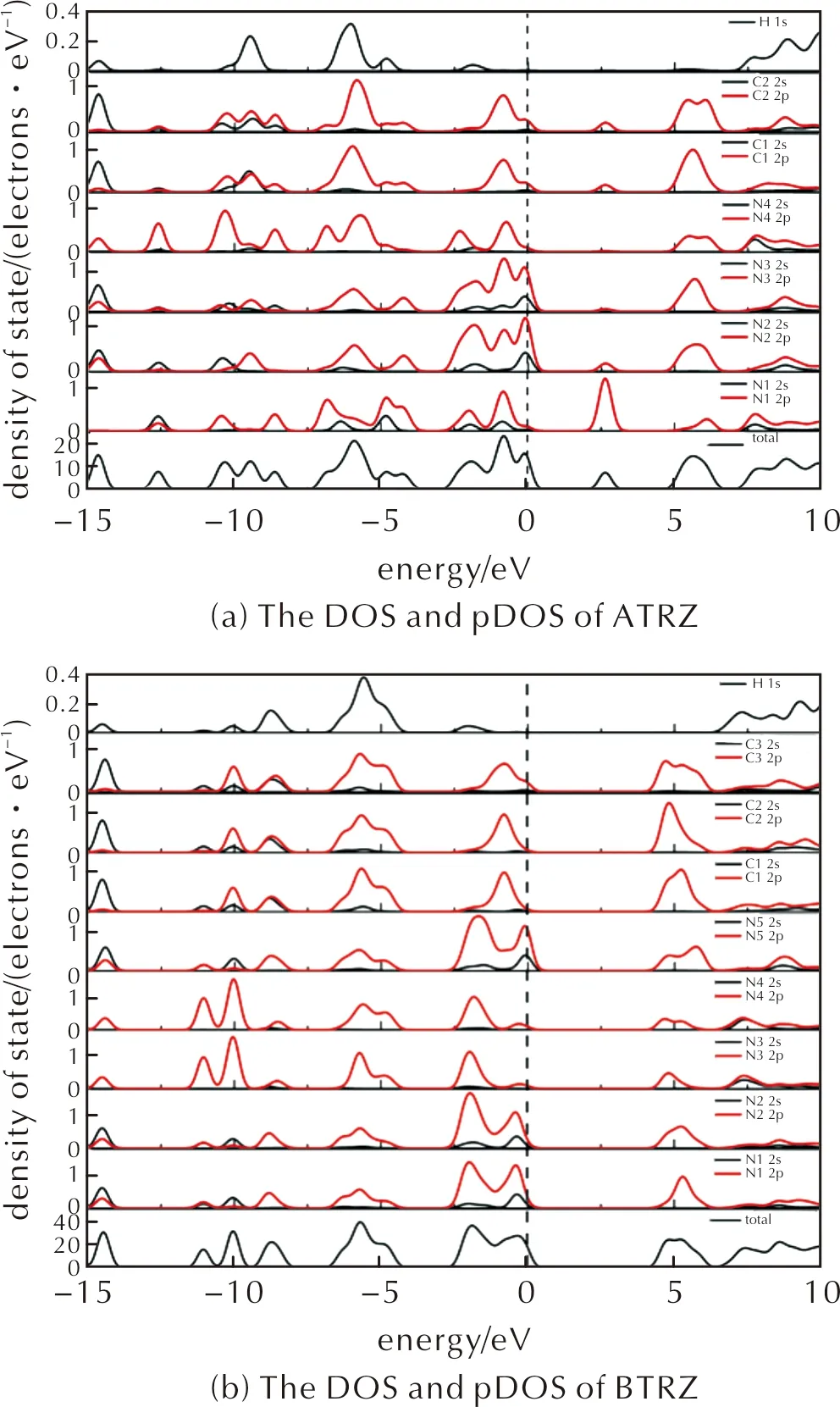
图5 ATRZ和BTRZ的总态密度和分态密度(DOS和pDOS)
在优化晶体结构的基础上,用GGA-PBE泛函计算了ATRZ和BTRZ的总态密度和分态密度(DOS和PDOS)如图5所示。ATRZ的带隙为2.51 eV。其中,-15~-13 eV能量区域中,能带主要由C/N的2s和2p态组成,对应C2和C5的sp2杂化电子与N的p电子。在-10~-3 eV区域中,主要由C/N的2p态和H的1s态组成,N的2s态贡献很少,主要对应于C2—N1/C5—N1键和C—H键;在费米能级附近,主要由C/N的2p态组成,并存在少量N的2s态,意味着C—N键和N—N键的高反应活性,也对应着N3原子的Lewis碱性。
BTRZ的带隙为5.0 eV,表明其具有绝缘性质,包含4组峰区。在最低能区-15~-14 eV附近,主要由C/N的2s和N的2p态组成,还有极少部分的H 1s态组成,对应于C—H键和C—N/C=N键;在-13~-4 eV附近,主要由C/N的2p态组成,还有少部分C的2s态组成,对应π型C—N键;在费米能级附近主要由C/N的2p态组成,还有少部分N的2s态,同样意味着C—N键和N—N键,以及N3/N4原子的孤电子对的高反应活性。
2.3 分子动力学模拟
基于ATRZ和BTRZ的晶体优化结构,本研究使用CPMD方法研究其在高温条件下(2 000 K)的初始分解机理,结果如图6和图7所示。
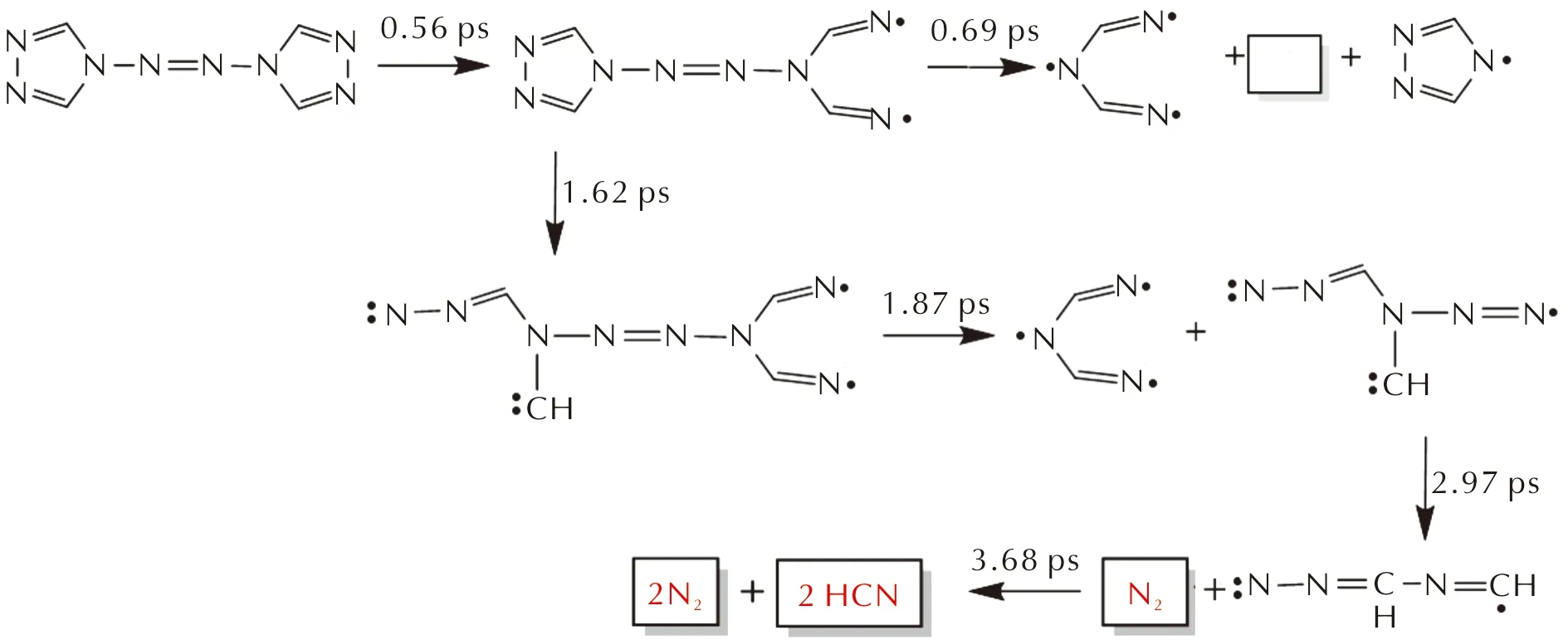
图6 凝固态ATRZ的分解机理(分解时间标在箭头上)
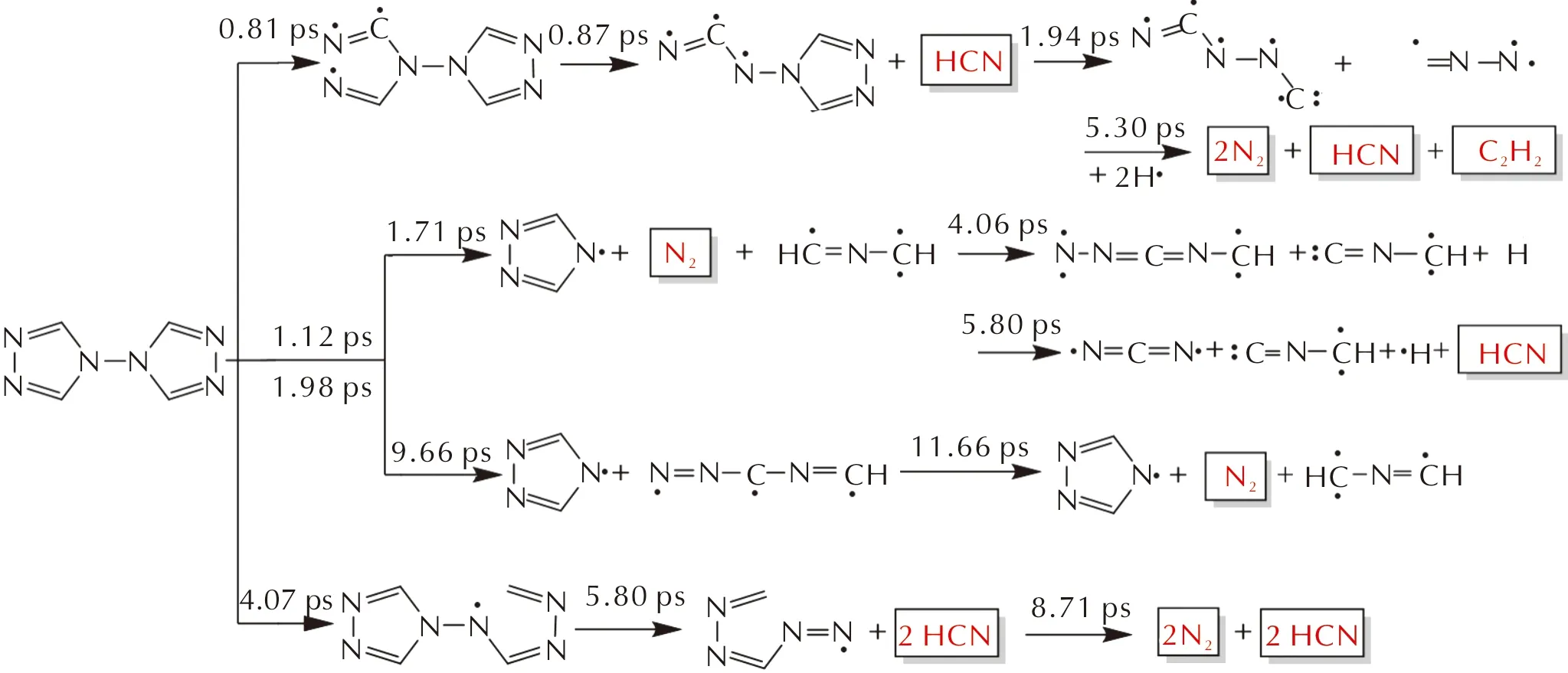
图7 凝固态BTRZ的分解机理(分解时间标在箭头上)
ATRZ的初始分解始于高温下N—N键的高频热振动,如图6所示,由于N原子上的孤电子对之间的斥力,环上的N3—N4键首先打开,进而引发键级最小的C1—N2键断裂。C1—N2键断裂意味着ATRZ的分解进入链增长阶段,包含两个路径:一种是几乎与N—N键协同发生的,在0.69 ps处N4—N6和N7—N11键同时断裂,生成气体产物N2;另一种是1.62 ps时第二个五元环上C—N键的断裂,并在2.97 ps处生成气体产物N2,最终3.68 ps处生成气体产物HCN和N2。
BTRZ晶体包含3种分解机理,如图7所示,分别为N1—N2键的高频振动开环(0.81 ps),几乎协同发生键级最小的C—N键的断裂并产生HCN,随着5.3 ps的自由基淬灭,第一种反应路径产生产物N2、HCN以及C2H2。第二种反应路径源于N1—N8键的断裂,链增长阶段包括两种不同的开环方式,一种为C=N双键的断裂,产生N2;另一条反应路径为C—N键断裂,与第一条反应路径类似。第三种反应路径为BTRZ不经过N1—N2键的振动开环引发,直接发生最弱C—N键断裂。第二种和第三种反应产物均为N2与HCN。
3 结 论
(1)基于部分三唑、四唑类化合物生成热DFT计算值与实验值的对比,表明TPSS/6-311++(3d,2p)方法得到的计算值与实验值最为接近。因此,该理论方法适用于三唑、四唑类化合物的分子结构优化和热力学性质的计算。
(2)ATRZ的爆轰参数显著大于BTRZ,这一现象可归因于ATRZ中偶氮键的引入使得三唑环之间的共轭程度增加,大大提高了分子的生成焓。
(3)分子体系的键长、Wiberg键级、ADCH电荷和静电势分布,均对应了其相应的晶体结构特征。费米能级处的态密度表明三唑环中高活性的C—N与N—N单键均可作为良好的配体与金属发生配位。动力学分解路径也进一步表明,ATRZ和BTRZ的分解引发端对应三唑环的高活性位点C—N与N—N单键,各反应路径均始于N—N键的断裂或N—N/C—N键的协同断裂,产物均包含HCN与N2。
猜你喜欢
成都大学学报(自然科学版)(2021年1期)2021-05-22 01:31:18
火工品(2019年6期)2019-06-05 02:35:44
厦门大学学报(自然科学版)(2018年2期)2018-04-11 07:07:35
当代化工研究(2016年5期)2016-03-20 16:21:32
中国果菜(2016年9期)2016-03-01 01:28:41
中国粮油学报(2016年1期)2016-02-06 02:17:09
化工进展(2015年6期)2015-11-13 00:26:50
西华师范大学学报(自然科学版)(2015年3期)2015-02-27 15:31:19
火炸药学报(2014年5期)2014-03-20 13:17:47
中国蔬菜(2014年3期)2014-02-01 18:06:17
