
贵金属(Pt、Pd和Ru)基催化剂在含氯挥发性有机物催化降解中的研究进展
2023-06-13 13:02赵亚若田明姣
能源环境保护 2023年3期
杨 沁, 徐 晗, 赵亚若, 田明姣, 何 炽
(西安交通大学 能源与动力工程学院 环境科学与工程系, 陕西 西安 710049)
0 引 言
含氯挥发性有机物(CVOCs)普遍有较高的毒性和致癌、致畸作用,是一类严重威胁人类和生物健康、破坏自然生态环境的污染物[1]。CVOCs治理技术通常包括直接焚烧、水蒸气重整、催化加氢脱氯、光催化氧化、催化降解等。其中,催化降解法由于具有能耗低、效率高、二次污染少等显著优点被认为是最有效可行的CVOCs脱除方法之一[2]。Pt、Ru、Pd等贵金属基催化剂尽管成本较高,但其在低温条件下对碳氢化合物分子具有较高的活化能力,在VOCs催化氧化领域得到了广泛的研究和应用[3-6]。在CVOCs氧化反应中,贵金属在低温下易与Cl作用,覆盖/惰化活性位,导致催化剂低温活性下降[7-9]。这一不足使贵金属催化剂在CVOCs降解中的应用受到较大限制。因此,开发具有高效和优异抗氯性的贵金属催化剂对工业CVOCs减排控制至关重要。从1993年开始,贵金属基催化剂用于CVOCs催化降解的研究逐步展开,经过一系列发展,贵金属基催化剂催化CVOCs体系逐步迈向成熟(图1)。
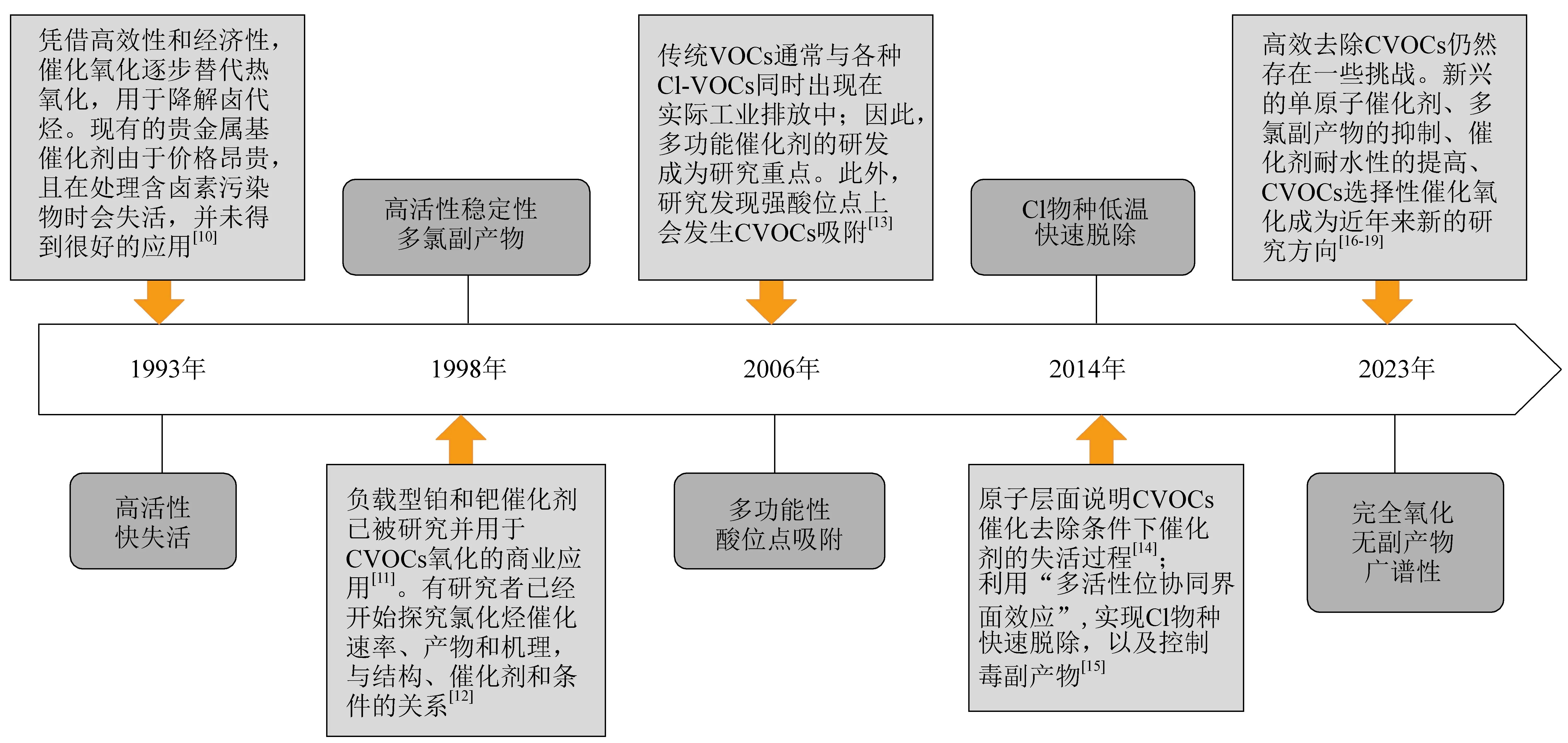
图1 贵金属基催化剂用于CVOCs催化降解发展年份图Fig. 1 Developing of noble metal based catalysts for CVOCs catalytic degradation
目前,CVOCs催化降解催化剂主要以Al2O3、TiO2、MgO、CeO2等单一或复合金属氧化物为载体,以Pt、Pd、Ru等为反应活性中心[20]。负载型贵金属催化剂的催化性能受到许多因素的制约和影响,如活性中心属性(负载量、颗粒尺寸、分散度等)及其与其他金属助剂/载体的相互作用。常用的提升负载型贵金属催化剂CVOCs催化性能的方式有金属助剂掺杂、活性中心状态优化、载体性质调控、反应条件调节等[1]。
1 Pt基催化剂
在贵金属基催化剂中,Pt基催化剂具有优异的低温可还原性能,对各种非卤代VOCs表现出优异的催化性能,已成功实现商业化推广应用[3-6]。然而,在CVOCs的催化氧化过程中,有机碳骨架的氯化会产生大量的多氯代副产物[7-9],且Pt基催化剂容易遭受氯攻击和碳沉积导致严重失活,因此开发具有良好活性和抗氯能力的Pt基催化剂对于有效控制CVOCs排放具有重要意义。针对Pt基催化剂易失活、生成多氯副产物等问题,研究者往往通过金属改性、载体性质调控、Pt化学状态优化等方法加以改进,使其催化性得到了较大提升。
1.1 金属间协同作用
负载型双金属催化剂通常表现出较单组分催化剂更加优异的催化性能,体现出独特的金属协同作用[3, 20-21]。单一Pt负载型催化剂可以增强原始载体的活性和多功能性,但同时会促进多氯代副产物和Cl2的生成,通过添加另一种金属修饰的方法可以有效抑制多氯代副产物的产生,提高催化剂的耐久性[22]。
Su等[23]合成了Pt-Co/HZSM-5三元催化剂用于催化降解二氯甲烷(DCM),在仅添加微量Pt(质量分数0.01%)的情况下,90% DCM的催化降解温度(T90)为249 ℃,比0.01Pt/HZSM-5和20Co/HZSM-5催化剂分别低67 ℃和42 ℃(图2(a))。研究发现,由于Co(以Co3O4的形式存在)对Pt原子的有效锚定实现了Pt的单原子分散,高分散Pt中心增加了Co3O4表面氧空位的比例,不仅使0.01Pt-Co/HZSM-5催化剂具有较好的反应活性,其主要脱氯副产物(CH3Cl)的最大选择性也在180 ℃时由7.5%降低至3.1%(图2(b))。
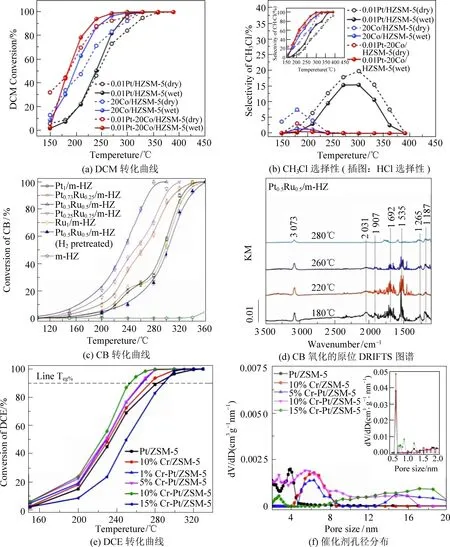
图2 负载型双金属催化剂的CVOCs催化降解性能[23-25]Fig. 2 Catalytic oxidation of CVOCs over supported bimetallic catalysts[23-25]
Wang等[24]采用改进的共浸渍法制备了系列不同Pt/Ru比的PtxRuy/m-HZ催化剂,Pt和Ru纳米粒子以Pt-O-Ru结构高度分散在多级孔HZSM-5载体上,这种结构提高了催化剂的氧化还原性和酸性,从而使得催化剂具有较高的抗Cl能力及更好的氯苯(CB)催化活性。Pt0.5Ru0.5/m-HZ催化剂上50% CB的催化降解温度(T50)仅为234 ℃,远低于Pt1/m-HZ和Ru1/m-HZ催化剂(T50≈300 ℃)(图2(c))。基于原位红外(insituDRIFTS)结果(图2(d)),研究者提出了Pt0.5Ru0.5/m-HZ催化剂上CB的氧化脱除强化机理:(1)增强CB在HZSM-5多级微介孔骨架上的吸附能力;(2)通过亲核取代将CB中的Cl提取至Brønsted酸位,形成HCl和酚盐;(3)氧化还原能力和酸性的协同,实现了HCl的低温脱除;(4)表面活性氧物种对酚盐的攻击形成短链烃类;(5)短链烃深度氧化为最终产物(COx、HCl和H2O)。相应地,催化剂的氧化还原行为、酸性以及提供的表面氧参与了Pt-O-Ru界面的协同作用,从而起到促进CB氧化的作用。
Shi等[25]的研究表明,适量Cr物种的掺杂可以促进Pt和Cr物种之间的强相互作用,有利于形成更多的Cr6+物种和酸性位点,强氧化位点(Cr6+/Pt物种)和酸性位点的协同作用有利于1,2-二氯乙烷(DCE)的脱氯以及中间产物和副产物的深度氧化。其中10%Cr-Pt/ZSM-5催化剂的T90低至255 ℃,在DCE的降解过程中只能观察到CO2的产生,并且具有良好的稳定性,在50 h的连续反应中催化剂上DCE转化率仅从95%略微下降到89%(图2(e))。但过量的金属引入也有可能破坏载体结构或引发活性物种团聚,导致催化性能降低(图2(f))。
1.2 载体的影响与作用
在CVOCs催化氧化中,载体属性在负载型Pt催化剂的催化性能及其稳定性中起着关键作用,其影响主要取决于载体的本征性质和Pt的存在形式[26]。一般来说,催化剂的载体可分为两类[27]:一种是活性载体,其本身对CVOCs的氧化具有一定的催化活性,如CeO2、Co3O4、MnOx等[28-29];另一种是惰性载体,如C、CaCO3等,通常为活性组分的均匀分散提供表面和孔结构,但其本身对CVOCs的消除没有催化活性[30-32]。目前的研究致力于合成具有适当物化属性的载体,以改善Pt在CVOCs氧化过程中易发生氯中毒、积碳等问题(图3)。
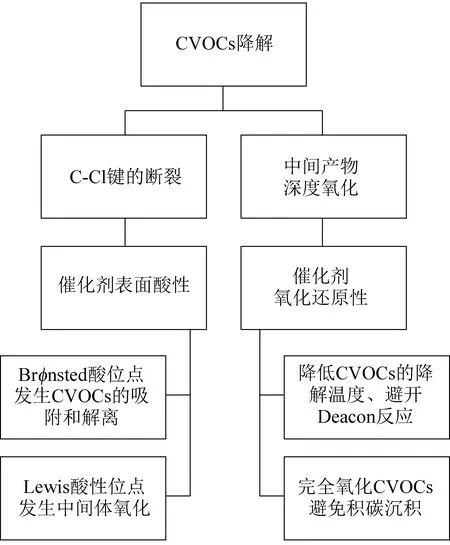
图3 CVOCs催化剂载体作用Fig. 3 Support roles of CVOCs catalysts
纳米结构多孔材料由于具有高的比表面积和丰富的孔隙,常被用作催化剂载体以分散活性中心,如He等[33]制备了纳米结构介孔CuO-MnO-CeO催化剂用以探究其在CB降解中的催化行为及协同作用。此外,纯纳米结构的铬也用作贵金属和过渡金属氧化物催化剂的催化活性载体,得益于铂颗粒所在的铬表面区域发生了协同作用,该催化剂在DCE和CB的催化氧化中表现出优异的性能[34]。0.5%(质量分数)Pt的引入使CrOOH催化剂在320 ℃脱除DCE的比速率常数提高了2.5倍(从2.1×10-3g·m-2·h-1到5.3×10-3g·m-2·h-1),将50%和90%DCE转化所需温度降低了20 ℃(图4(a))。0.5%Pt/CrOOH催化剂上,氯苯在380 ℃实现了完全氧化,CO2选择性>95%,而负载型Cr2O3/γ-Al2O3催化剂在完全氧化氯苯时CO2选择性仅为90%;与CrOOH和15%Cr2O3/Al2O3相比,0.5%Pt/CrOOH催化剂的T50和T90分别降低了20 ℃和40 ℃。
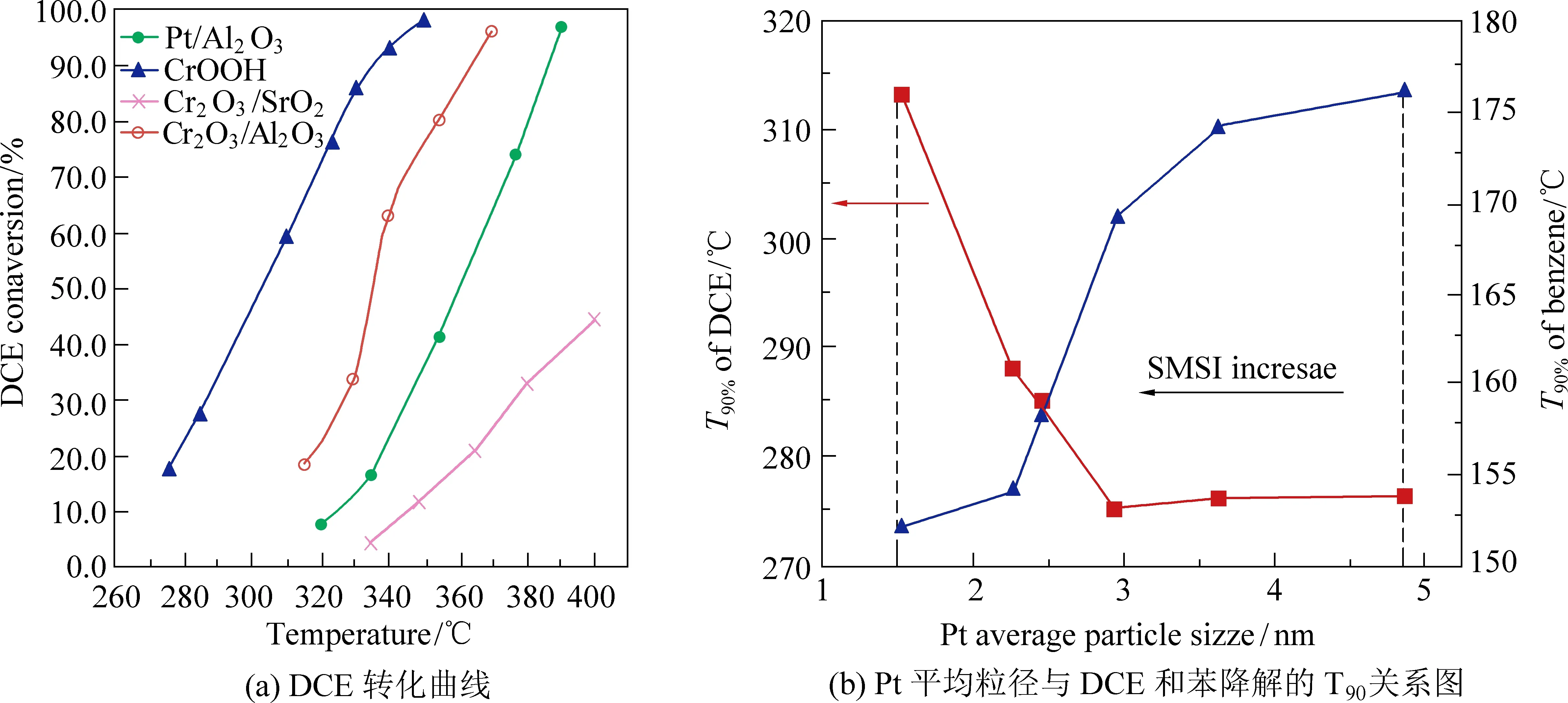
图4 Pt/CrOOH和Pt/CeO2-TiO2的DCE催化降解性能[34, 36]Fig. 4 Catalytic oxidation of DCE over Pt/CrOOHand Pt/CeO2-TiO2[34, 36]
由金属间化合物纳米晶构建的具有原子尺度金属/氧化物界面的双功能催化剂具有优异的抗氯中毒能力[35]。Shi等[36]采用乙二醇还原法制备了系列Pt纳米颗粒尺寸可控的Pt/CeO2-TiO2催化剂用于CVOCs低温去除,发现Pt粒径减小增强了PtOx物种与CeO2-TiO2复合氧化物的金属-载体相互作用(图4(b))。得益于Pt纳米颗粒尺寸效应及其与载体间的作用,Pt/CeO2-TiO2催化剂在DCE催化转化中表现出优异的催化活性和反应稳定性。值得注意的是,Pt/CeTi-9催化剂对苯和DCE的降解均表现出优异的催化活性,T90分别只有154 ℃和288 ℃,远低于文献报道的大多数催化剂,并且在DCE催化降解中,C2H3Cl是唯一副产物。
载体表面酸性和氧化还原性也密切影响着催化剂的催化性能,有利于CVOCs的化学吸附和氧物种活化[37]。例如,SnO2上的Brønsted酸位可以为HCl的生成提供充足的质子,有利于消除吸附在催化剂表面的Cl物种[35]。具有高酸性和较强还原能力的Pt/Al2O3已被报道对DCM氧化具有活性,在420 ℃下DCM转化率为100%、HCl产率为92%[38]。另一项工作中,在Pt/γ-Al2O3上实现了440 ℃下氯苯的完全氧化[7]。然而,负载型Pt催化剂易因碳物种的沉积和氯中毒导致失活[39]:一方面,碳和氯物种的沉积可以覆盖DCM分解的活性位点;另一方面,氯物种的吸附导致Pt物种失活。为了进一步提高CVOCs催化降解中Pt催化剂的HCl或Cl2活性和选择性,需要考虑Pt和载体的化学状态。
CeO2具有丰富的氧空位和可逆Ce3+/Ce4+变价能力,它不仅可以作为贵金属的载体,还可以作为吸附和活化氧物种的关键活性位点[40-41]。凭借其独特的物理化学性质,CeO2在CVOCs的氧化反应中得到了广泛研究[42-43]。当Pt分散在CeO2上时,Pt将与CeO2发生电子相互作用,促进催化反应进行。Pt-CeO2之间的强相互作用可以促进中间产物的深度氧化、抑制碳沉积,从而提高催化剂降解CVOCs的耐久性[36, 44]。Zhang等[45]通过控制CeO2的形貌,利用Pt与CeO2之间的相互作用来调节Pt的化学态,进而影响氯乙烯(VC)燃烧的催化性能。Pt/CeO2八面体催化剂被证实由于Pt和CeO2之间的弱相互作用形成了PtO2从而具有较强的VC吸附和断裂C—Cl键的能力。在VC燃烧中,CeO2-R的表现优于CeO2-C和CeO2-O;400 ℃时,50%的VC在CeO2-R上完成转化,而在CeO2-O上仅有30%VC完成转化;负载Pt显著促进了VC分解,尤其是CeO2-O(T90=280 ℃)。
1.3 其他因素
除载体种类之外,催化反应条件,如CVOCs的种类和浓度、气体流速、催化剂用量及空速等对CVOCs催化降解也有重要影响[46]。催化剂的选择性高度依赖于VOCs的种类。例如,贵金属负载型催化剂通常对脂肪烃、含氧VOCs和芳香烃的燃烧具有优异的催化活性和稳定性,但这些催化剂不适合催化氧化含Cl-和S-的VOCs,主要原因是Cl-、S-导致催化剂中毒[47-48]。研究表明,甲苯、正己烷、乙酮、丙酮等VOCs的存在可以提高Pt-Pd基催化剂上三氯乙烯(TCE)和三氯甲烷(TCM)的转化率[49]。
CVOCs中一些无机组分的加入也会对催化剂的催化效果产生影响。如臭氧的加入可以提高Pt/Al2O3上1,2-二氯乙烷的转化率[46]。而水蒸气的存在可以显著提高Pt/Al2O3催化三氯乙烯(TCE)、四氯乙烯(PCE)过程中对HCl的选择性,提升了TCE和PCE的低温(400 ℃以下)氧化速率[50-51]。以水作为供氢反应物,可以显著提高催化剂对HCl的选择性,7 500 ppm的水使Pt/Al2O3的HCl产率从37.5%增加到58.9%。
2 Pd基催化剂
用于CVOCs氧化的催化剂的活性组分主要由过渡金属氧化物(CoOx、VOx、CrOx和MnOx)和贵金属(Pt、Ru和Pd)组成,通常负载在氧化铝、沸石和二氧化铈等载体上。尽管过渡金属氧化物成本低廉,但由于形成易挥发金属氧氯化物,造成活性相的流失,故而其应用受到限制。与金属氧化物相比,贵金属表现出更高的抗挥发性和催化活性。在众多关于贵金属催化剂中,负载Pd催化剂凭借其优异的高活性和热稳定性,得到广泛研究[52]。
氧化铝负载Pt、Pd催化剂在催化氧化三氯乙烯(TCE)反应中,Pd/Al2O3的T50为400 ℃,而Pt/Al2O3的T50为435 ℃,Pd/Al2O3表现出更优异的催化活性[53]。在高于400 ℃时,Deacon反应(HCl+O2H2O+Cl2)促进了Cl2的形成。Pd比Pt更倾向于产生氯气,且Pd基催化剂在催化CVOCs过程中,易产生大量多氯代有机副产物。如常用商业催化剂(Pd/γ-Al2O3、Pd/ZSM-5和Pd/SiO2)催化氧化1,2-二氯苯(1,2-DCB),在氧化过程中,均能检测到大量的三氯苯、四氯苯和五氯苯等多氯代有机副产物。反应过程中Pd的氯化程度可能是形成多氯有机副产物的原因,主要是由于活性氧与氯化钯反应生成次氯酸盐物种,该物种可以通过亲电取代继续与吸附的氯酚物种反应(图5),形成多氯有机副产物[54]。研究者进一步探究了Pd/ZSM-5(25)催化剂上不同氧气和水含量对1,2-二氯苯(1,2-DCB)催化氧化的影响,发现降低氧气含量和增加水含量均可以导致1,2-DCB催化活性的提高,同时多氯代有机副产物的数量和种类会减少。更重要的是,在厌氧条件下,当以水为唯一氧化剂时,Pd/ZSM-5(25)催化剂上1,2-DCB完全催化降解的温度与好氧条件相同为500 ℃,并且副产物只有氯苯和1,3-二氯苯[55]。
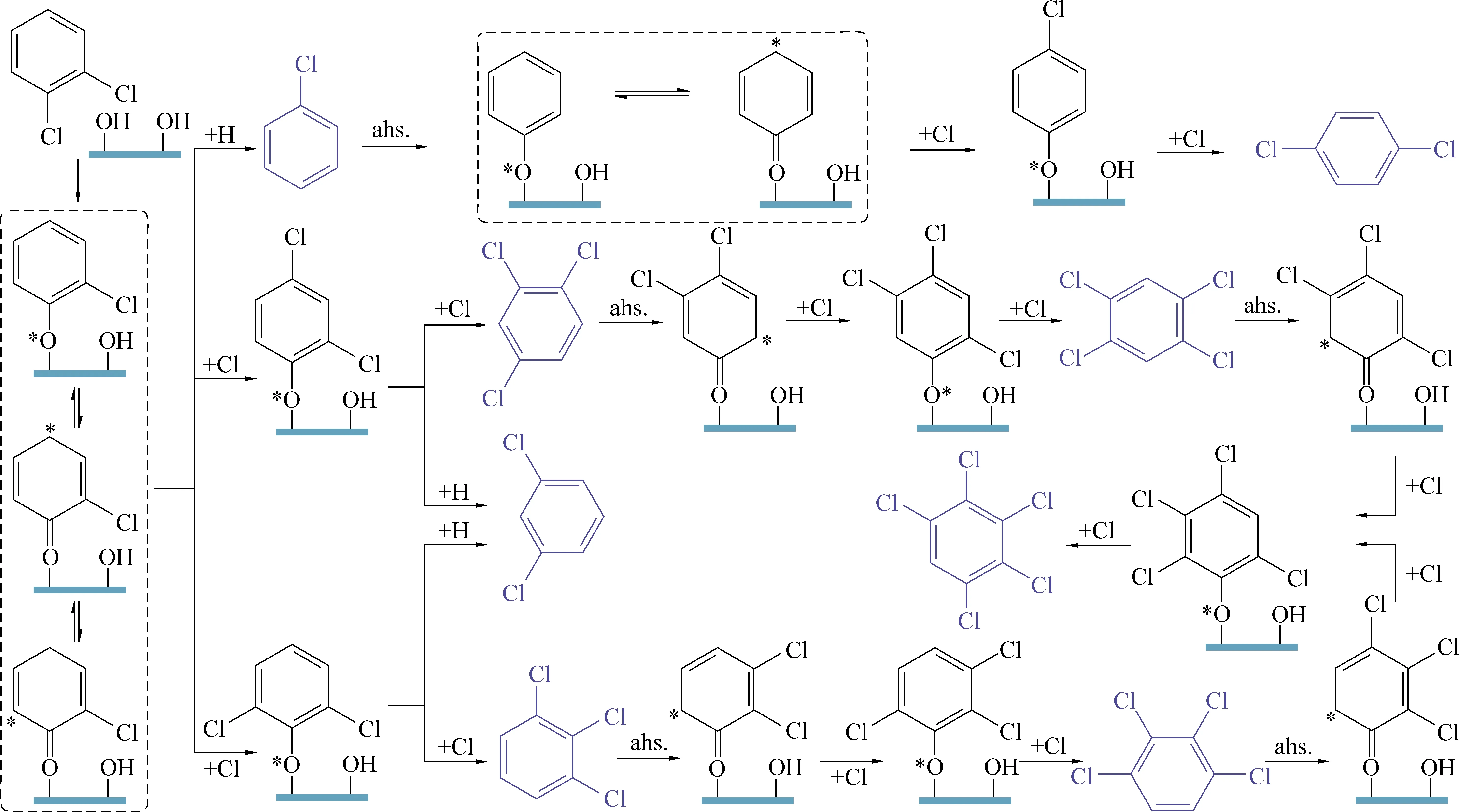
图5 多氯代有机副产物的形成机理[54]Fig. 5 Proposed mechanism for the formation of polychlorinated organic by-products[54]
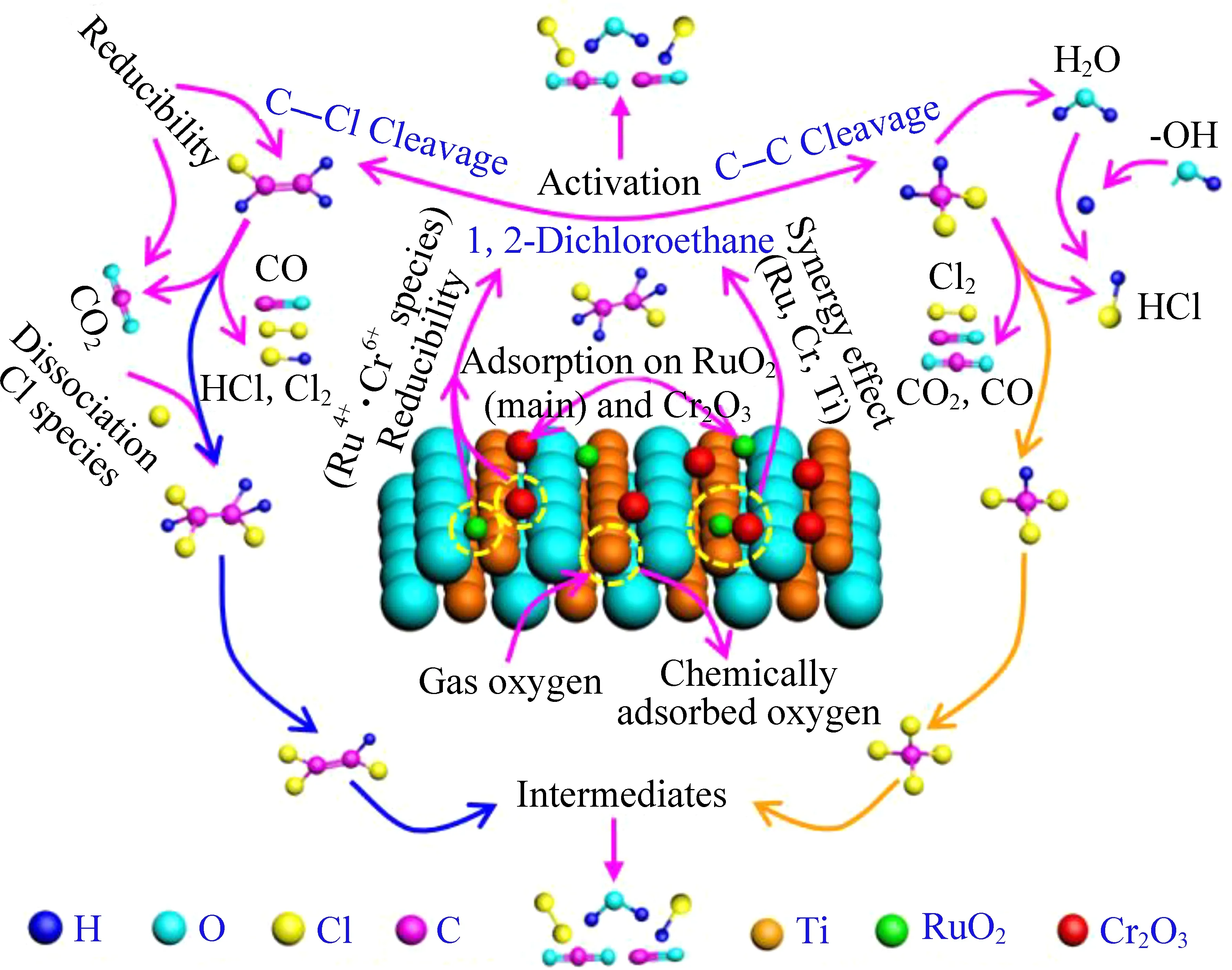
图6 Cr改性的RuO2/TiO2催化剂对 1,2-二氯乙烷的降解机理[73]Fig. 6 Proposed 1,2-dichloroethane destruction mechanism over Cr-modified RuO2/TiO2 catalyst[73]
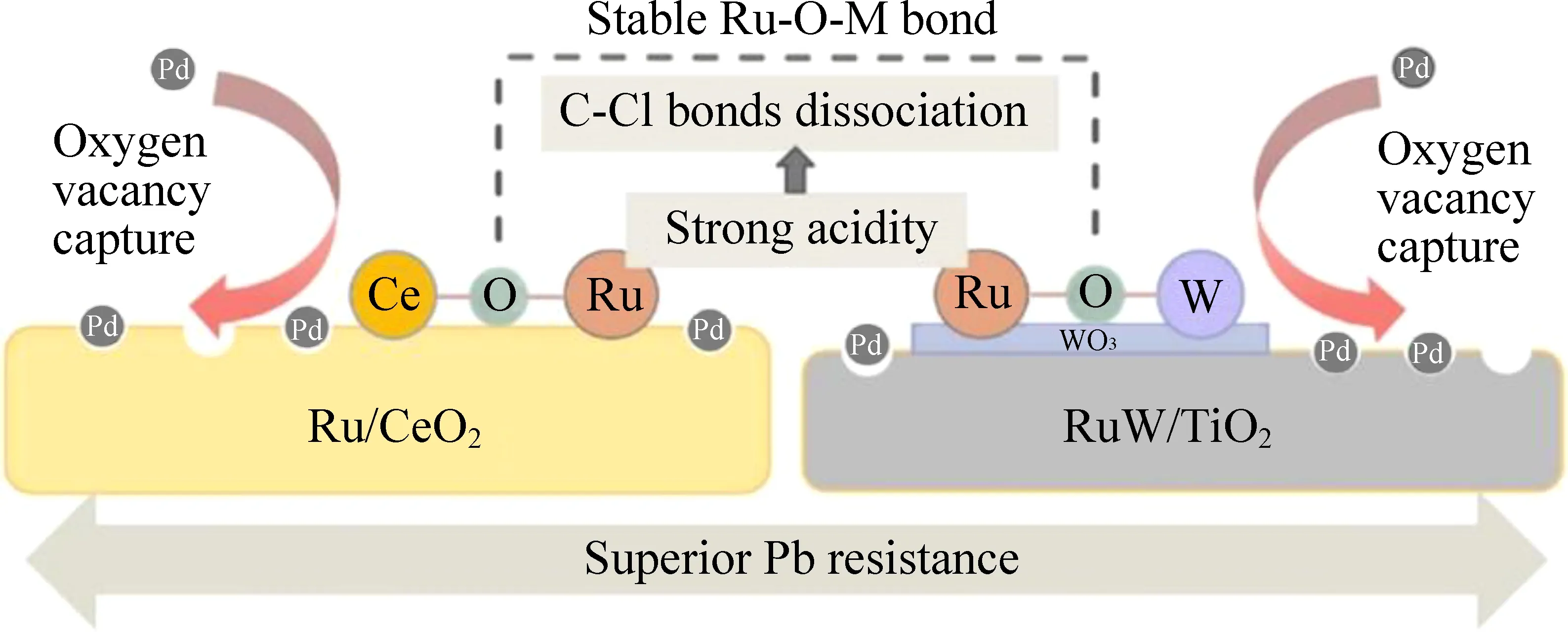
图7 Ru/CeO2和RuW/TiO2催化剂的抗铅机理示意图[89]Fig. 7 Schematic illustration for Pb resistant mechanism of Ru/CeO2 and RuW/TiO2 catalysts[89]
目前,烧结、氯吸附和碳沉积是导致Pd催化剂失活的主要原因,可以通过改变载体的性质来提高催化剂的催化性能和稳定性。如通过使用氧化铝、沸石、氧化铈、ZrO2、TiO2等作为载体来分散Pd物种并产生新的相互作用以达到增强其活性、选择性和稳定性的目的[51, 56-58]。有研究者比较了不同La基钙钛矿LaBO3(B=Co、Mn、Fe、Ni)负载的预还原钯催化剂在空气中对氯苯全氧化的催化性能,结果表明Pd/LaFeO3在氧化氯苯和耐氯性中表现出最佳的综合性能[59]。此外,0.93Ru2.87Pd/3DOM CeO2催化剂得益于RuPd纳米粒子与3DOM CeO2之间形成紧密的纳米界面[60],具有优异的吸附氧量、低温还原性以及TCE吸附活化能力。催化剂上路易斯酸位点越强,产生的有机副产物就越少[58],因为其表面酸中心可以作为卤代烃的有效化学吸附中心。有研究合成了羟基磷灰石(HAP)用以负载Pd并应用于DCE氧化反应,得益于HAP适度的酸度及其与Pd活性相的相互作用,在反应过程中没有生成中间产物氯乙烯[52]。
3 Ru基催化剂
与Pt和Pd相比,Ru基催化剂的研究相对较少。近年来,Ru基催化剂因其在烷烃、芳香烃尤其是卤代VOCs的催化氧化中表现出优异的催化性能而备受关注[62-64]。在CVOCs氧化反应中,Ru催化剂产生的多氯代副产物明显少于Pd和Pt基材料[39, 65-66]。Ru催化剂上通过Deacon反应[65, 67]可以高效地将氯物种从催化剂表面去除,但Cl2通过亲电取代与有机骨架反应,仍会有少量多氯代副产物生成。因此,研究者通过掺杂其他金属、改变载体种类状态、引入水蒸气等方式来提高Ru基催化剂的催化性能。
3.1 金属间协同作用
在以往的研究中,Ru的负载量在1%~5%(质量分数)之间[45, 64, 67]。考虑到该催化剂工业应用的成本,人们希望在尽量减少贵金属使用的同时保证催化剂的高活性、选择性和稳定性。在催化剂中添加其他金属氧化物来提高其综合性能是目前的研究热点,CeO2由于其独特的晶体结构、丰富的氧空位和优异的储氧能力受到了广泛的关注[67-68]。密度泛函理论(DFT)计算证实,当氧化铈掺杂其他过渡金属(Cu、Mn、Fe、Ni)或贵金属(Pd、Pt、Ru)时,氧空位形成能会降低,更容易从掺杂剂和氧化铈[69-70]中释放氧气。
在Ru-Ce/TiO2催化剂催化降解CB中,CeO2与Ru物种相互作用形成Ru-O-Ce,促进电子转移,提高表面氧的迁移率,从而形成价态更高的RuOx[71]。与0.4Ru/TiO2催化剂相比,0.4Ru-1.0Ce/TiO2催化剂的T10和T90分别降低了50 ℃和60 ℃。在170 ℃时,0.4Ru/TiO2催化剂的反应速率从1.21×10-8mol/(min·m2)提高到10.45×10-8mol/(min·m2),优于2.0Ru/TiO2催化剂。Ce的引入提高了COx产率和对无机氯(HCl和Cl2)的选择性,阻止了催化剂产生更多的氯代副产物,降低了二噁英的产生几率。
RuO2的负载通过丰富表面氧物种可以极大地增强催化剂的氧化还原性能,促进深度氧化过程,提高了产物对CO2的选择性;V2O5的负载可以提升TiO2的酸性,有利于DCM的吸附和C—Cl键的断裂,从而提高其低温催化活性。因此,将氧化中心(RuO2)与酸中心(V2O5)耦合的RuO2-V2O5/TiO2催化剂在DCM分解中具有良好的应用前景[72]。研究表明,钒的加入可以促进DCM向CO的转化而不是CH3Cl,RuO2负载在TiO2上可以增强CO向CO2的深度氧化。
Huang等[73]制备了Cr改性的RuO2/TiO2催化剂,并用于1,2-DCE降解。Cr2O3和RuO2/TiO2的协同作用增强了表面Ru的暴露,产生了丰富的可还原Cr6+和Ru4+物种和化学吸附氧,显著提高了催化剂的1,2-DCE降解活性和CO2/HCl选择性。Cr2O3负载后的RuCrTi催化剂的初始还原温度由RuTi催化剂的145 ℃降低为128 ℃。研究发现,1,2-DCE主要在RuO2/Cr2O3/TiO2的Lewis酸位(LAS)上活化,发生C—Cl键断裂;同时,C—C断裂伴随着脱HCl和氯化反应,Cr2O3的存在大大提高了RuO2/TiO2的LAS浓度和氧化还原能力,加速了1,2-DCE的深度破坏,抑制了CH2Cl2、C2HCl3和CHCl3的生成(图 6)。
Mn、Sn、W等过渡金属的掺杂同样可以诱导Ru负载的催化剂产生丰富的活性氧物种,提升氧化还原能力、氧迁移率和表面酸性,从而提高对CVOC的反应性能[74-76]。Hu等[73]采用等体积浸渍法制备了Ru-Mn/TiO2催化剂用于CB的催化氧化。其中0.8%Ru-10%Mn/TiO2催化剂在250 ℃时CB转化率为99%,CO2产率在300 ℃时为95%。Ru物种提高了Mn4+和Ru4+的浓度,降低了H2还原温度,有利于提高催化剂的氧化还原性能,而Ru/Mn界面处Ru和Mn物种之间的相互作用促进了氧空位和表面活性氧物种的形成,进而增强了催化活性。
3.2 载体的影响与作用
Ru在催化氧化CVOCs的过程中可发生Deacon反应,促进材料表面的氯物种快速脱附,加速CVOCs在过渡金属氧化物上的氧化。Ru/Al2O3、Ru/Co3O4和Ru/CeO2对1,2-二氯乙烷、二氯苯和氯苯的氧化具有较高的稳定活性[65, 77-78]。然而,反应产生的Cl2并不是一种环境友好的产物,而且催化氧化过程中Cl2在催化剂表面的强吸附容易导致催化剂的失活,氯原子可以阻塞催化活性位点,从而使催化剂因中毒失效。因此,从催化剂表面去除氯物种是关键步骤。通常Cl的解离与吸附发生在Lewis酸性位点上,而来自Bronsted酸的质子可以促进Cl以HCl的形式脱除。因此,如果能结合Ru与Cl物种稳定的氧化活性以及载体较强的Bronsted和Lewis酸性,就可以获得对CVOCs具有高催化活性和高HCl选择性的催化剂。
Wang等[79]报道了在MCF表面生长纳米结构的Co3(PO4)2(记为CoPO)的异质结构CoPO-MCF介孔杂化材料的合成路径。介孔载体提供了大的比表面积,较强的酸性和还原性,在此基础上,引入Ru前驱体制得的Ru/CoPO-MCF催化剂在低温转化氯乙烯(VC)中表现出优异的催化性能。研究发现由于纳米磷酸钴具有的独特性质,CoPO-MCF载体与钌物种的相互作用比纯MCF或块状CoPO更强。这些CoPO纳米相生长在二氧化硅表面上,形成独特的互穿异质结构。Ru物种在这些CoPO纳米相上的负载可以通过Ru载体相互作用的异质性诱导。此外,由于Ru物种与CoPO-MCF载体之间的相互作用,1%Ru/CoPO-MCF催化剂氧化还原能力得到增强。其次,CoPO-MCF载体比其他载体具有更强的酸性,当反应温度不足以将VC完全氧化为CO2、H2O和HCl时,它可以有效抑制Cl物种的吸附和氯化副产物的产生。这些原因使得1%Ru/CoPO-MCF对VC氧化的催化活性要比1%Ru/MCF和1%Ru/CoPO更好(T50=278 ℃;T90=313 ℃)。当反应温度升高至400 ℃时,1%Ru/CoPO-MCF几乎完全氧化了CHCl3,而1%Ru/MCF仍有1 ppm的CHCl3产生,且副产物CH2Cl2的量远低于1%Ru-MCF。

研究表明,RuCrTi[73]、Ru/Pi-CeO2[84]、Ru/3DOM SnO2[85]均能长期高效降解CVOCs,适应不同的水汽环境。然而催化剂的实际应用环境往往比较复杂,烟气中通常含有多种重金属,包括铅(Pb)、镉(Cd)、砷(As)等,这些重金属会沉积在催化剂表面,从而导致催化剂性能明显下降[86-88]。载体不仅在一定程度上能提高Ru的低温可还原性能和催化活性,同时可有效地提升其抗重金属中毒能力。Lv等[89]系统地研究了CVOCs氧化过程中Pb中毒对不同Ru基催化剂活性的影响(图 7)。结果表明载体(CeO2或TiO2)上的氧空位可以捕获Pb并将其牢牢锁定在晶格中,表现出强捕获效应;此外,稳定的Ru-O-M(M=Ce和或W)键的形成削弱了活性Ru位点与Pb之间的亲和力,保证了Pb优先沉积在氧空位处。在上述因素的协同作用下,Ru/CeO2和RuW/TiO2催化剂中Ru位点的本征活性(以强酸性为主)得以保留,保证了C—Cl键的高效解离和CB的氧化。
贵金属颗粒的分散程度同样是决定其催化性能的关键。因此,设计改性贵金属催化剂去除CVOCs的一个思路就是如何改善活性组分与载体之间的分散性,载体与活性组分之间的相互作用也可以改善催化剂的催化性能[70]。Zhang等[90]比较了多孔载体(Al2O3、MgO、微孔HZSM-5)对双金属RuCo纳米颗粒(NPs)催化氧化1,2-DCE性能的影响。RuCo/HZSM-5具有较强的氧化还原能力、适宜的酸性、良好的1,2-DCE吸附能力、高度分散的RuCo NPs以及RuCo NPs与HZSM-5之间较强的相互作用,表现出最佳的抗Cl性能和催化活性。与之类似,研究者开发了不同Ru前驱体活性位点调控的RuOx/Sn0.2Ti0.8O2催化剂,以二氯甲烷为模型分子,活性测试结果表明,使用Ru胶体优化Ru前驱体显著提高了催化剂的活性(当Ru负载量为1%时,T90降低了约90 ℃)[91]。研究发现,由于活性物种分散度的提高(Ru团簇尺寸从3~4 nm减小到约1.3 nm)以及活性物种与载体之间相互作用的增强,不仅使得TOF值提高了近一倍,提高了活性组分的利用效率,同时也增强了催化剂的整体氧化性能。
3.3 其他因素
水在CVOCs的氧化过程中起着重要的作用,通常归因于以下三点:(1)由于清洗作用,氯和碳物种得到有效去除[92]。因为催化剂比氯物种容易吸附更多的OH,所以水产生的H+优先与催化剂表面的Cl-结合形成HCl[93];(2)碱性OH的增加是由于从水中吸附了更多的O原子[94],这可以促进OH与1,2-DCE分子中Cl原子的相互作用,并且OH有利于氧气在MOx催化剂上的吸附和转移[95];(3)在水存在的条件下,Deacon过程容易发生逆反应,提高了HCl的选择性[96]。
一般来说,水的引入往往会导致反应活性的降低,这是由于水和氧气或反应物分子在活性中心的竞争吸附造成的。也有研究者提出水的引入减少了中强酸的量,特别是Brønsted酸位点的量[97]。研究发现,在Ru/WO3和Ru/TiO2催化剂催化氧化1,2-DCE的过程中,水蒸气的引入产生的OOH物种会转化为更多的活性氧(O2-和O-)物种,水直接促进了氯乙烯向CH3COOH的转化;此外,水可以部分吸附在Ru/WO3样品表面的羟基上,为1,2-DCE分子提供吸附位点[98]。
Ru基催化剂在潮湿条件下可以显著促进氯物种的脱附,有效抑制有害多氯副产物的生成[91]。H2O的引入可以阻断部分反应途径,抑制C2Cl4的生成[97],促进Cl与羟基的交换反应,从而有利于HCl的生成,并通过C2H3Cl→醇→醛→羧酸的反应过程减少其他氯物种或Cl2的生成[99]。
4 总结与展望
根据VOCs的特性来合理设计开发催化剂是实现其工业规模应用的重要方向。在CVOCs中,无机氯物种易于分离且吸附力强容易导致催化剂快速失活。从活性部位快速去除无机氯通常从以下两个途径出发:(1)通过Ru、Mn等金属的掺杂将游离氯氧化为氯气从体系中脱除;(2)引入Bronsted酸位点,将游离氯转化为HCl避免催化剂氯中毒失活。实现CVOCs高效降解的催化剂应具有出色的氧化还原能力以提高CO2选择性,同时含有丰富的表面酸位点以抑制多氯代副产物的形成,进而提高HCl选择性和抗Cl中毒能力。
本文综述了铂族金属(Pt、Pd和Ru)催化降解CVOCs的研究进展,总结和比较了不同增强催化性能的技术手段,如与其他金属协同、载体性质调变、水蒸气引入等;阐述了反应CO2选择性和副产物的生成可以通过选择合适的催化剂或反应条件(温度、湿度和气体组分)进行优化,为CVOCs催化降解高效催化剂的设计提供了参考。贵金属负载型催化剂具有优异的活性和稳定性,但同时也存在成本高、易氯中毒、易因碳沉积而失活等问题,阻碍其实际工业应用。尽管目前已经有了大量针对不同种类CVOCs催化降解的研究,但高效去除CVOCs仍然存在一些挑战。开发具有成本效益的催化剂并最大限度地利用金属活性位点是非常必要的。新兴的单原子催化剂是一种可能的解决方案,亟待进行深入的环境应用研究。此外,多氯副产物的抑制和催化剂耐水性的提高也需进一步探究。此外,目前大多数研究致力于将CVOCs完全氧化为H2O、CO2和HCl,这种方法虽然可以避免CVOCs对环境的毒害作用,但同时也会造成一定的资源浪费。将CVOCs选择性催化氧化,使目标产物资源化利用成为近年来新的研究方向。
猜你喜欢
分子催化(2022年1期)2022-11-02
贵金属(2021年1期)2021-07-26
贵金属(2021年1期)2021-07-26
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26
保鲜与加工(2021年1期)2021-02-06
广东饲料(2016年5期)2016-12-01
中国资源综合利用(2016年12期)2016-01-22
淮南师范学院学报(2015年3期)2015-03-22
河北科技大学学报(2015年5期)2015-03-11
应用化工(2014年12期)2014-08-16
