
经典名方三化汤基准样品量值传递研究
2023-06-08 12:21刘明松邓亚伟忻晓东梁董茜李春花刘晓明
中草药 2023年11期
刘明松,邓亚伟,忻晓东,梁董茜,李春花,2*,刘晓明
1. 河北中医学院药学院,河北 石家庄 050200
2. 河北省中药组方制剂技术创新中心,河北 石家庄 050090
3. 河北省药品审评中心,河北 石家庄 050090
三化汤(Sanhua Decoction)为国家中医药管理局公布的《古代经典名方目录(第一批)》第55 首,来源于金·刘完素《素问病机气宜保命集》卷中[1]。原文记载:“中风外有六经之形证,先以加减续命汤,随证治之,内有便溺之阻格,复以三化汤主之。”可见三化汤治疗中风临床应用已久[2]。全方由大黄、枳实、羌活、厚朴4 味药组成,君药大黄与枳实、厚朴3 药合用,泻热祛瘀、通腹降气,再加羌活祛风解表,有升有降、表里结合,共治内风外风,是中医治疗中风的经典方[3-5]。
目前,有关三化汤的研究多集中于临床、药理和含量测定等方面,对其量值传递研究较少[6]。在国家药品监督管理局药品审评中心组织起草的《按古代经典名方目录管理的中药复方制剂药学研究技术指导原则(试行)》要求中明确指出,经典名方基准样品的关键质量属性(critical quality attributes,CQAs)的研究应包括特征图谱、指标成分含量及干膏率等关键信息[7-8]。本研究在文献考证结合煎煮工艺结果的基础上确定三化汤制备工艺,制备了15 批基准样品煎液,通过构建其特征图谱并对指标性成分进行含量测定,确定其含量及转移率范围,并结合出膏率分析饮片-基准样品量值传递规律,为其质量研究提供科学依据,也为三化汤中药复方制剂进一步研究提供参考。
1 仪器与材料
1.1 仪器
Waters H-Class 型超高效液相色谱仪、Empower工作站,美国Waters 公司;YP2002 型电子天平,上海津平科学仪器有限公司;D5.5-L 型煎药壶,潮州市潮安区康雅顺电器有限公司;Milli-QReference超纯水系统,法国Millipore 公司;KQ500D 型超声波清洗器,昆山市超声仪器有限公司;HCP-1000A型高速多功能粉碎机,浙江省永康市金穗机械制造厂;HWS-28 型恒温水浴锅、DHG-9030A 型电热鼓风干燥箱,上海一恒科学仪器有限公司。
1.2 试药
对照品厚朴酚(批号PS012117,质量分数≥98.5%)、紫花前胡苷(批号PS010688,质量分数≥99.5%)、紫丁香苷(批号PS012100,质量分数99.86%)、柚皮苷(批号PS012062,质量分数≥99.5%)、新橙皮苷(批号PS011239,质量分数99.54%)、橙皮苷(批号 PS011588,质量分数99.54%)、芸香柚皮苷(PS012543,质量分数≥98.5%),均购自成都普斯生物科技股份有限公司;对照品大黄酚(批号20120320)、芦荟大黄素(批号20120320),质量分数均≥98%,购自上海源叶生物科技有限公司;对照品和厚朴酚(批号MUST-20032205,质量分数>98%)、大黄酸(批号MUST-11032801,质量分数>98%)、大黄素(批号MUST-19100810,质量分数≥98%),购自成都曼思特生物科技有限公司。甲醇,色谱级,赛默飞世尔科技有限公司;甲酸,色谱级,天津大茂化学试剂厂;三氯甲烷,天津大茂化学试剂厂;乙腈,色谱级,天津市康科德科技有限公司。
不同产地及批号饮片见表1,经河北中医学院药学院段绪红副教授鉴定,大黄为蓼科大黄属植物药用大黄RheumofficinaleBaill.的干燥根和根茎,枳实为芸香科柑橘属植物酸橙CitrusaurantiumL.的干燥幼果,羌活为伞形科羌活属植物羌活NotopterygiumincisumTing ex H. T. Chang 的干燥根茎和根,厚朴为木兰科厚朴属植物厚朴Magnolia officinalisRehd. et Wils.的干燥干皮及枝皮,以上均为符合《中国药典》2020 年版炮制要求的饮片。通过随机数表法对表1 中不同批次三化汤饮片进行随机组合,结果见表2。

表1 三化汤饮片来源信息Table 1 Source information of Sanhua Decoction herbal pieces

表2 三化汤随机组合Table 2 Random combination table of Sanhua Decoction
2 方法与结果
2.1 三化汤基准样品的制备
三化汤原文记载“每服三两,水三升,煎至一升半,终日服之。”经文献考证,确定处方中各饮片所用剂量及加水量,最终明确煎煮工艺。确定全方为厚朴、大黄、枳实、羌活各30 g,粉碎过10 目筛,加纯水2100 mL,浸泡0.5 h,开盖,武火煮沸后,保持文火(500 W)微沸状态约90 min,200 目滤网滤过,滤液放凉,调整体积至1050 mL,即得三化汤基准样品。同法制备三化汤各单味饮片及缺大黄、缺枳实、缺厚朴、缺羌活的阴性样品溶液。
2.2 三化汤基准样品特征图谱的建立
2.2.1 色谱条件 色谱柱为Acquity UPLC®BEH C18柱(100 mm×2.1 mm,1.7 μm);流动相为乙腈-0.1%甲酸水溶液,梯度洗脱:0~5 min,10%~13%乙腈;5~11 min,13%~17%乙腈;11~31 min,17%~25%乙腈;31~32 min,25%~29%乙腈;32~40 min,29%~34%乙腈;40~43 min,34%~47%乙腈;43~55 min,47%~75%乙腈;体积流量0.2 mL/min;进样体积2 μL;柱温35 ℃;检测波长255 nm。三化汤基准样品UPLC 图见图1-A,混合对照品图见图1-B。
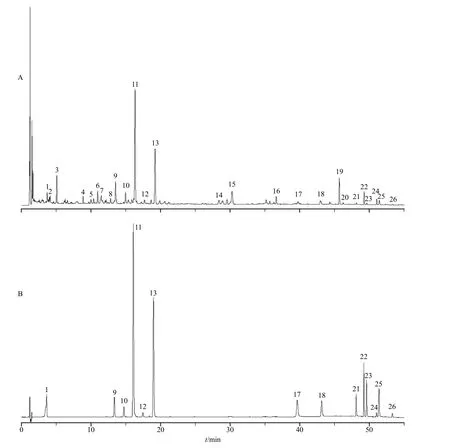
图1 三化汤基准样品的UPLC 图谱 (A) 和对照品图谱 (B)Fig. 1 UPLC chromatogram (A) and comparator chromatogram (B) of substance benchmarks in Sanhua Decoction
2.2.2 供试品溶液的制备 精密吸取“2.1”项制备的煎液2 mL,甲醇定容至10 mL 量瓶中,摇匀,静置,取上清液过0.22 μm 滤膜,即得供试品溶液。
2.2.3 对照品溶液的制备 分别取异欧前胡素、大黄酸、大黄素、大黄酚、和厚朴酚、厚朴酚、芦荟大黄素、大黄素甲醚、紫丁香苷、紫花前胡苷、柚皮苷、橙皮苷、芸香柚皮苷和新橙皮苷对照品适量,精密称定,甲醇定容,制成质量浓度为25.21、15.25、16.11、19.13、17.29、18.22、16.34、12.31、42.11、72.08、210.64、36.24、45.62、171.97 μg/mL 的混合对照品溶液。
2.2.4 精密度考察 取S1 号供试品溶液,按“2.2.2”项下方法制备,按“2.2.1”项下色谱条件连续进样6 次,色谱图中新橙皮苷(13 号峰)保留时间适中且峰形较好、分离度高,因此以新橙皮苷为参照峰,计算各共有峰相对保留时间和相对峰面积,结果显示各共有峰相对保留时间的RSD 均<0.5%,相对峰面积的RSD 均<1.5%,表明仪器精密度良好。
2.2.5 重复性考察 取同一批三化汤基准样品,平行制备6 份供试品溶液,按“2.2.1”项下色谱条件进样,以新橙皮苷(13 号峰)为参照峰,计算各共有峰相对保留时间和相对峰面积,结果显示各共有峰相对保留时间的RSD 均<0.8%,相对峰面积的RSD 均<1.8%,表明该方法重复性好。
2.2.6 稳定性考察 取同一供试品溶液,按“2.2.1”项下色谱条件,分别于制样后0、2、4、8、12、24 h 进样,以新橙皮苷(13 号峰)为参照峰,计算各共有峰相对保留时间和相对峰面积,结果显示,各共有峰相对保留时间的RSD 均<0.9%,相对峰面积的RSD 均<2.6%,表明供试品溶液在24 h 内稳定性良好。
2.3 三化汤特征图谱评价及饮片-基准样品量值传递关系研究
2.3.1 基准样品相似度评价 将表2 不同批次饮片组合按“2.2.2”项制备方法制备15 批三化汤基准样品供试品溶液,按“2.2.1”项色谱条件测定,记录色谱图。将S1~S15 批基准样品特征图谱导入“中药色谱指纹图谱相似度评价系统(2012 版)”生成叠加图(图2),以S1 为参照图谱,时间窗宽度0.1 s,设置中位数法,Marker 峰匹配,并生成对照图谱(R),S1~S15 批基准样品相似度结果分别为0.996、0.981、0.988、0.994、0.985、0.997、0.993、0.995、0.995、0.998、0.987、0.994、0.991、0.997、0.984,可见,各批次三化汤基准样品特征图谱相似度均大于0.98。
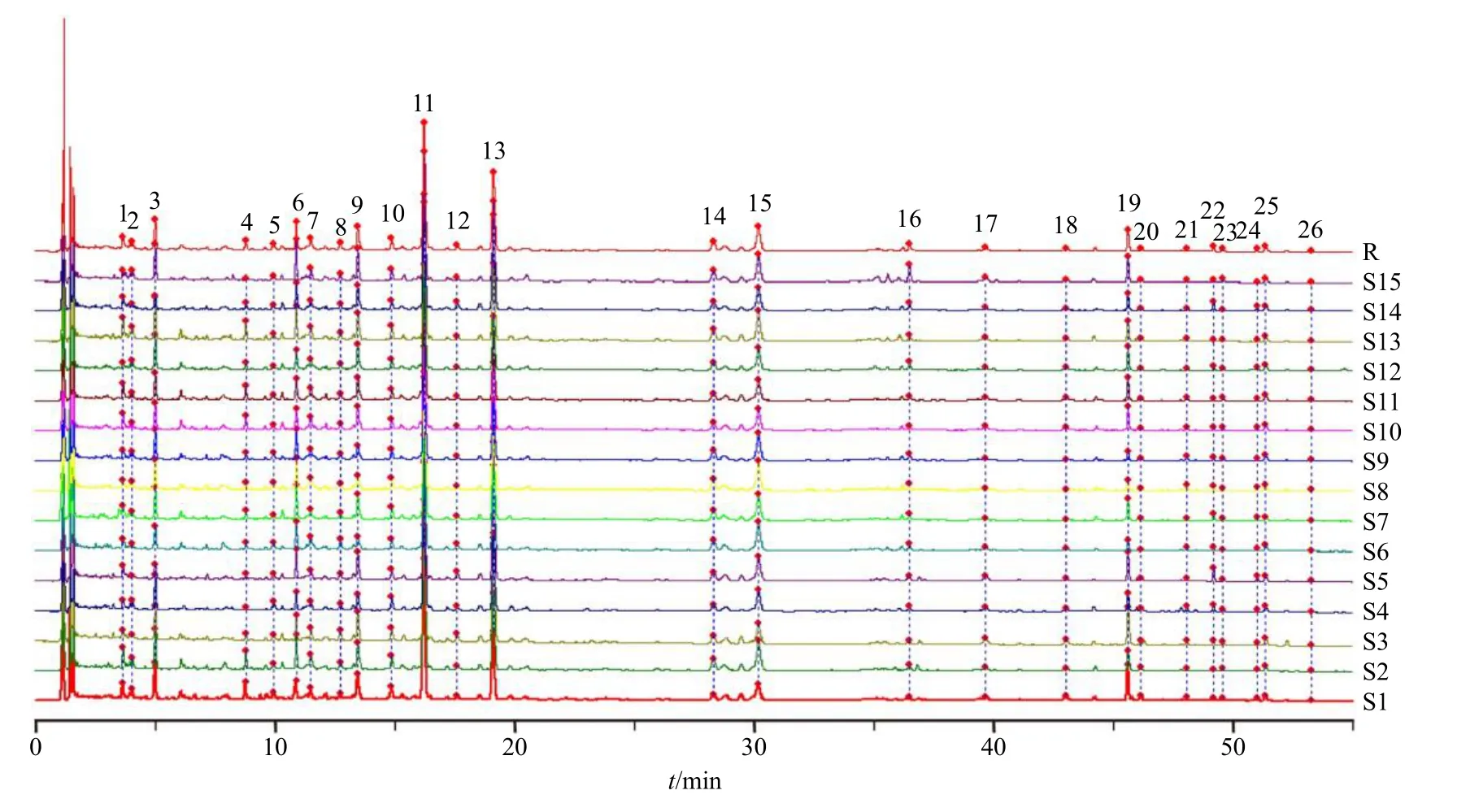
图2 15 批三化汤基准样品特征图谱Fig. 2 Characteristic chromatogram of substance benchmarks in 15 batches of Sanhua Decoction

图3 三化汤基准样品与单味饮片的UPLC 图谱Fig. 3 UPLC chromatogram of Sanhua Decoction benchmark samples and single herbal pieces
2.3.2 特征峰归属 按“2.2.2”项方法制备各单味饮片和阴性供试品溶液,按“2.2.1”项下色谱条件测定,记录色谱图。通过基准样品与各单味饮片(图3)及阴性供试品图谱(图4)比对,将各特征峰进行归属,全处方共归属26 个特征峰,其中峰6、7、14、15、16、17(芦荟大黄素)、18(大黄酸)、21(大黄素)、25(大黄酚)、26(大黄素甲醚)归属于大黄;峰1、2、4、22(和厚朴酚)、24(厚朴酚)归属于厚朴;峰3、8、10(芸香柚皮苷)、11(柚皮苷)、12(橙皮苷)、13(新橙皮苷)归属于枳实;峰5、9(紫花前胡苷)、19、20、23(异欧前胡素)归属于羌活。

图4 三化汤基准样品与阴性样品的UPLC 图谱Fig. 4 UPLC chromatogram of Sanhua Decoction benchmark samples and negative sample
2.3.3 量值传递关系研究 以每味药中的1 个特征峰为参照峰(大黄参照峰为15 号,枳实参照峰为13号,羌活参照峰为9 号,厚朴参照峰为22 号),对基准样品与各单味饮片共有峰峰面积比值作分析,结果见表3。各单味药与基准样品中对应共有峰峰面积虽存在差异,但大部分特征峰比值变化较小,结果表明各单味药特征图谱中主要物质成分可以从饮片-基准样品较为完整地传递,且各物质成分归属关系清晰。
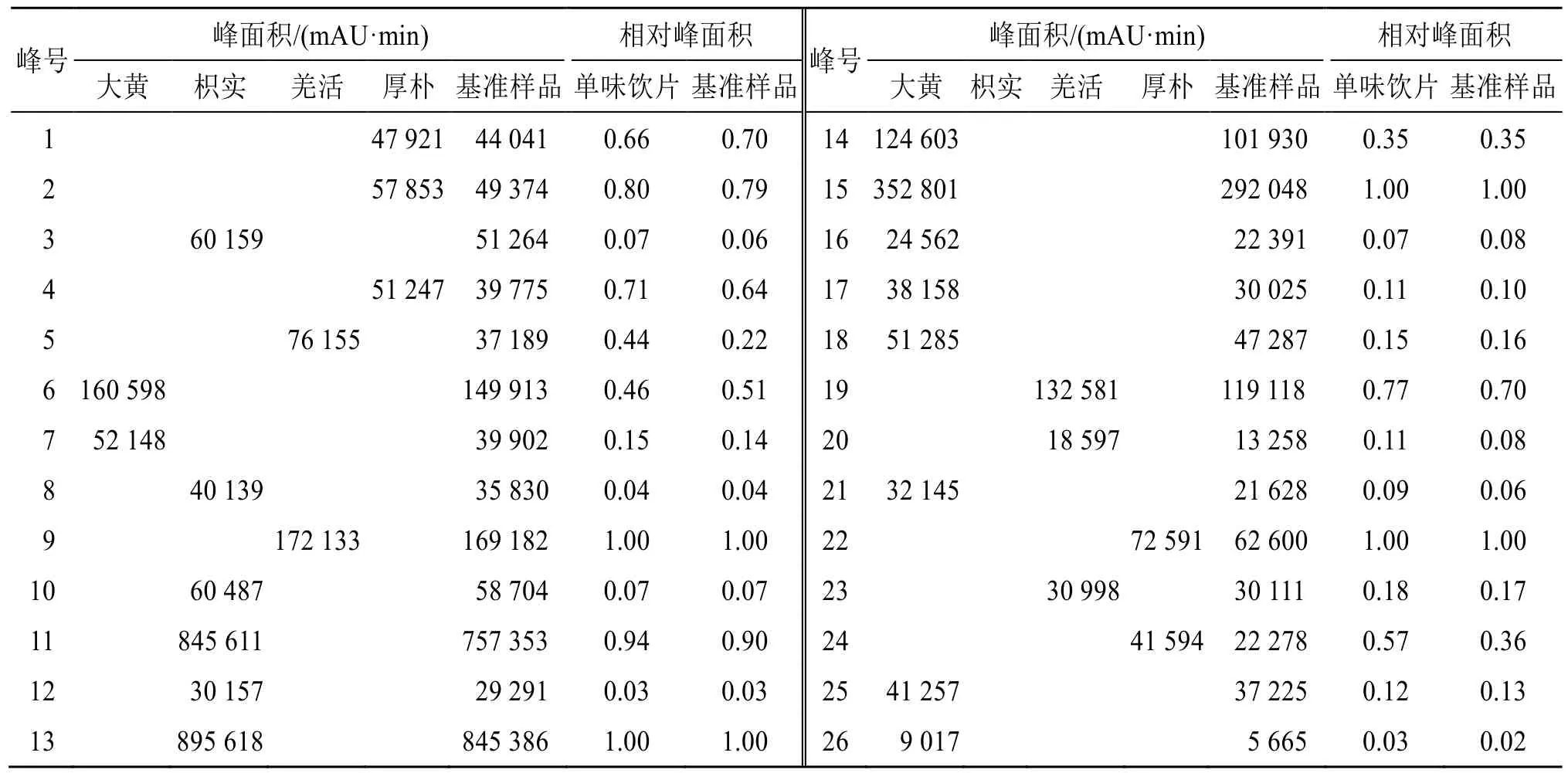
表3 三化汤基准样品与各药味饮片中特征峰峰面积比值Table 3 Peak area ratio of characteristic peaks between Sanhua Decoction benchmarks sample and each herbal pieces
2.4 三化汤基准样品及各单位饮片中指标成分含量测定方法的建立
2.4.1 色谱条件
(1)枳实饮片、羌活饮片、基准样品(紫花前胡苷、柚皮苷、新橙皮苷)UPLC 定量色谱条件:Acquity UPLC®BEH C18(100 mm×2.1 mm,1.7 μm)色谱柱;流动相为乙腈-0.1%甲酸水溶液,梯度洗脱:0~10 min,10%~17%乙腈;10~28 min,17%~26%乙腈;体积流量0.2 mL/min;进样体积2 μL;柱温35 ℃;检测波长290 nm。对照品见图5-A,基准样品见图5-B。

图5 混合对照品 (A) 和三化汤基准样品指标性成分 (B) 含量测定色谱图Fig. 5 Chromatogram for content determination of mixed control sample (A) and reference standard components of Sanhua Decoction (B)
(2)大黄饮片、厚朴饮片、基准样品(芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、和厚朴酚、厚朴酚)UPLC 定量色谱条件:Acquity UPLC®BEH C18(100 mm×2.1 mm,1.7 μm)色谱柱;流动相为乙腈-0.1%甲酸水溶液,梯度洗脱:0~8 min,30%~42%乙腈;8~18 min,42%~46%乙腈;18~22 min,46%~50%乙腈;22~30 min,50%~72%乙腈;体积流量0.2 mL/min;进样体积2 μL;柱温35 ℃;游离总蒽醌(芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚)检测波长254 nm,混合对照品色谱图见图5-A,基准样品见图5-B;和厚朴酚、厚朴酚检测波长295 nm,混合对照品色谱图见图5-A,基准样品见图5-B。
2.4.2 三化汤基准样品含量测定供试品溶液的制备(1)紫花前胡苷、柚皮苷、新橙皮苷:同“2.2.2”项下制法制备供试品溶液。
(2)游离总蒽醌、和厚朴酚、厚朴酚:精密量取基准样品汤液25 mL,置分液漏斗中,用三氯甲烷提取3 次,每次20 mL,合并三氯甲烷溶液,蒸干,残渣加适量甲醇溶解,转移至5 mL 量瓶中,加甲醇至刻度,摇匀,静置,过0.22 μm 滤膜,即得供试品溶液。
2.4.3 单味饮片含量测定溶液的制备
(1)枳实:取饮片粉末约0.1 g,精密称定,置圆底烧瓶中,精密加入甲醇30 mL,称定质量,加热回流1.5 h,放冷,再称定质量,用甲醇补足减失的质量,摇匀,静置,过0.22 μm 滤膜,即得。
(2)羌活:取饮片粉末约0.1 g,精密称定,分别置不同具塞锥形瓶中,加甲醇20 mL,超声处理20 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,过0.22 μm 滤膜,即得。
(3)大黄、厚朴:含量测定供试品溶液的制备参照《中国药典》2020 年版。
2.4.4 对照品溶液的制备 取紫花前胡苷、柚皮苷、新橙皮苷、芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、和厚朴酚、厚朴酚对照品适量,精密称定,甲醇溶解,制得质量浓度分别为248.01、908.02、992.01、68.42、86.01、64.41、74.80、32.41、204.01、156.80 μg/mL 的各对照品溶液。
2.4.5 线性关系考察 取不同质量浓度的指标性成分对照品溶液,采用“2.4.1”项下色谱条件进样测定,以质量浓度为横坐标(X),峰面积为纵坐标(Y),绘制标准曲线并计算回归方程,结果如表4 所示,结果表明芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、和厚朴酚、厚朴酚、紫花前胡苷、柚皮苷、新橙皮苷在各自的线性范围内线性关系良好。

表4 指标性成分含量测定方法学考察Table 4 Methodological investigation of determining of content of index components
2.4.6 精密度考察 取同一批三化汤基准样品(S1),参照“2.4.2”项下方法制备供试品溶液1 份,按“2.4.1”项下色谱条件,连续进样6 次,芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、和厚朴酚、厚朴酚、紫花前胡苷、柚皮苷、新橙皮苷峰面积的RSD 值均小于1.95%,RSD 见表4,表明仪器精密度良好。
2.4.7 重复性考察 取同一批三化汤基准样品,按“2.4.2”项下方法平行制备6 份供试品溶液,按“2.4.1”项下色谱条件进样检测,芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、和厚朴酚、厚朴酚、紫花前胡苷、柚皮苷、新橙皮苷质量分数的RSD 值均小于2.68%,RSD 见表4,表明该方法的重复性良好。
2.4.8 稳定性考察 取同一批三化汤基准样品(S1),参照“2.4.2”项下方法制备供试品溶液1 份,按“2.4.1”项下色谱条件,分别在制样后0、2、4、8、12、18、24 h 进行测定,芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、和厚朴酚、厚朴酚、紫花前胡苷、柚皮苷、新橙皮苷峰面积的RSD 见表4,结果表明供试品溶液在24 h 内稳定。
2.4.9 加样回收率考察 精密量取已测知指标成分含量的三化汤基准样品(S1)共6 份,分别按已知质量浓度的80%、100%和120%加入对照品,采用“2.4.2”项下方法制备供试品溶液,测定样品中各指标成分含量,计算加样回收率,结果如表4 所示,芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、和厚朴酚、厚朴酚、紫花前胡苷、柚皮苷、新橙皮苷的平均加样回收率均在95%~105%,RSD 均小于3.0%,表明方法准确度良好。
2.4.10 样品测定 取三化汤基准样品(S1~S15)和各单味药材饮片,分别按照“2.4.2”“2.4.3”项下方法制备供试品溶液,按照“2.4.1”项下色谱条件进样分析,计算三化汤基准样品和单味饮片中指标成分的含量。
2.5 三化汤饮片-基准样品中指标成分转移率考察
按照公式计算三化汤饮片-基准样品中指标成分的转移率。
w为基准样品中指标性成分的含量,W为饮片中指标性成分的含量
由表5、6 结果可知,15 批三化汤基准样品中指标成分紫花前胡苷质量分数在0.39%~0.72%,平均转移率为(28.08±5.28)%;柚皮苷质量分数在2.70%~3.77%,平均转移率为(45.07±5.17)%;新橙皮苷质量分数在1.69%~3.88%,平均转移率为(41.03±4.75)%;和厚朴酚与厚朴酚总质量分数在0.11%~0.32%,平均总转移率为(3.32±0.92)%;大黄游离总蒽醌质量分数在0.10%~0.28%,平均转移率为(16.54±3.57)%。
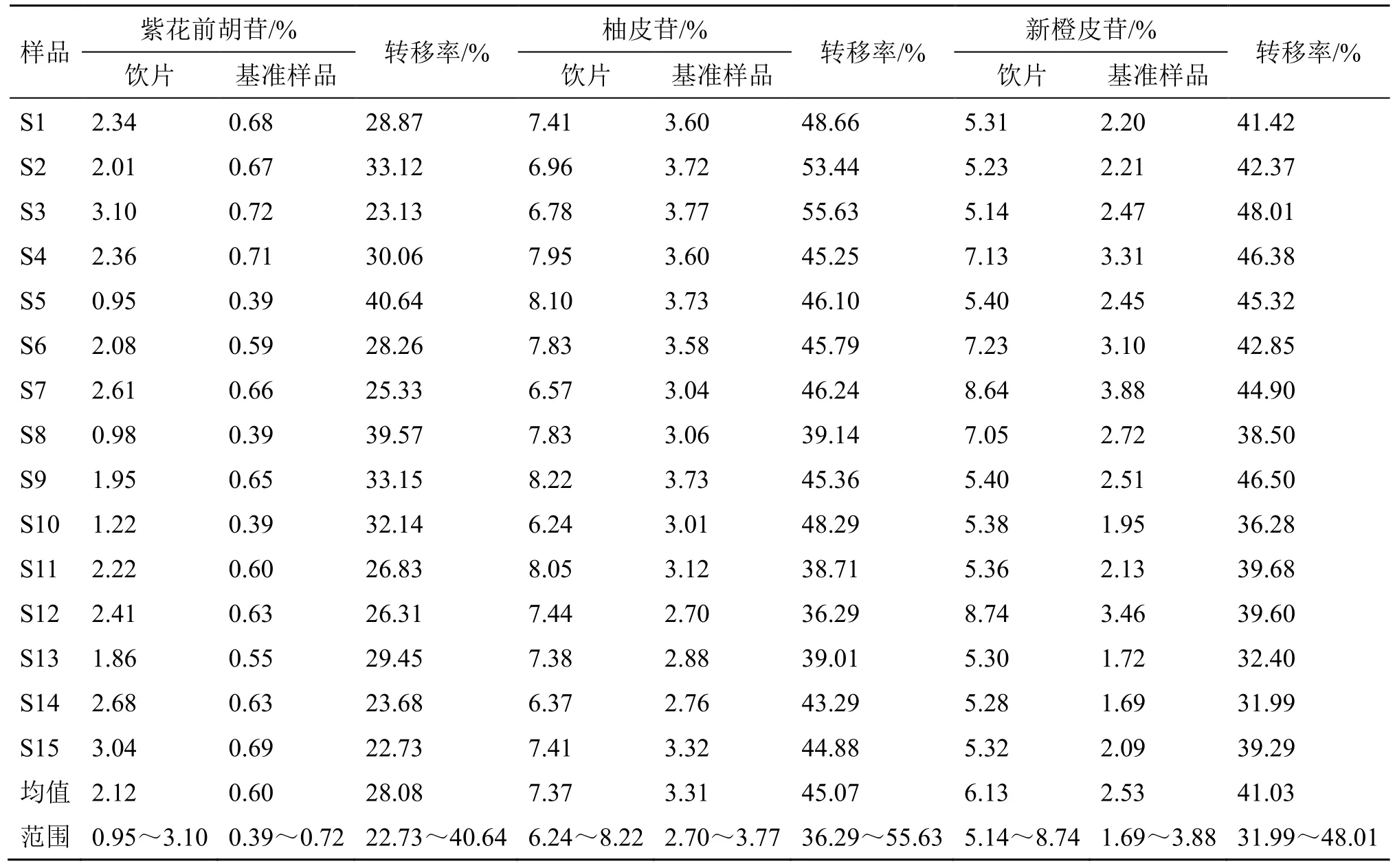
表5 三化汤饮片-基准样品紫花前胡苷、柚皮苷、新橙皮苷含量及转移率Table 5 Content and transfer rate of nodakenin, naringenin and neohesperidin in Sanhua Decoction herbal pieces-benchmark samples
2.6 出膏率测定
按“2.1”项下制备方法制备15 批基准样品汤液,精密吸取50 mL 于蒸发皿中,水浴蒸干,真空干燥48 h 后,置干燥器中至恒定质量,称定质量,计算得到各批次三化汤基准样品出膏率。
其中m表示干膏质量,M表示饮片质量,v代表取样体积,V代表汤液总体积
分别取15 批对应批号的大黄、枳实、羌活、厚朴单味饮片,按“2.1”项下方法制备得到4 味药单味饮片水煎液,按照上述制备方法,得到单味饮片出膏率。出膏率结果见表7。全处方共计120 g,则理论出膏率=大黄出膏率×25%+枳实出膏率×25%+羌活出膏率×25%+厚朴出膏率×25%,其中枳实出膏率最高(44.61%±3.01%),厚朴的出膏率最低(13.61±0.67)%,各批次处方干膏转移率为64.43%~76.22%,均保持在均值的±10%。
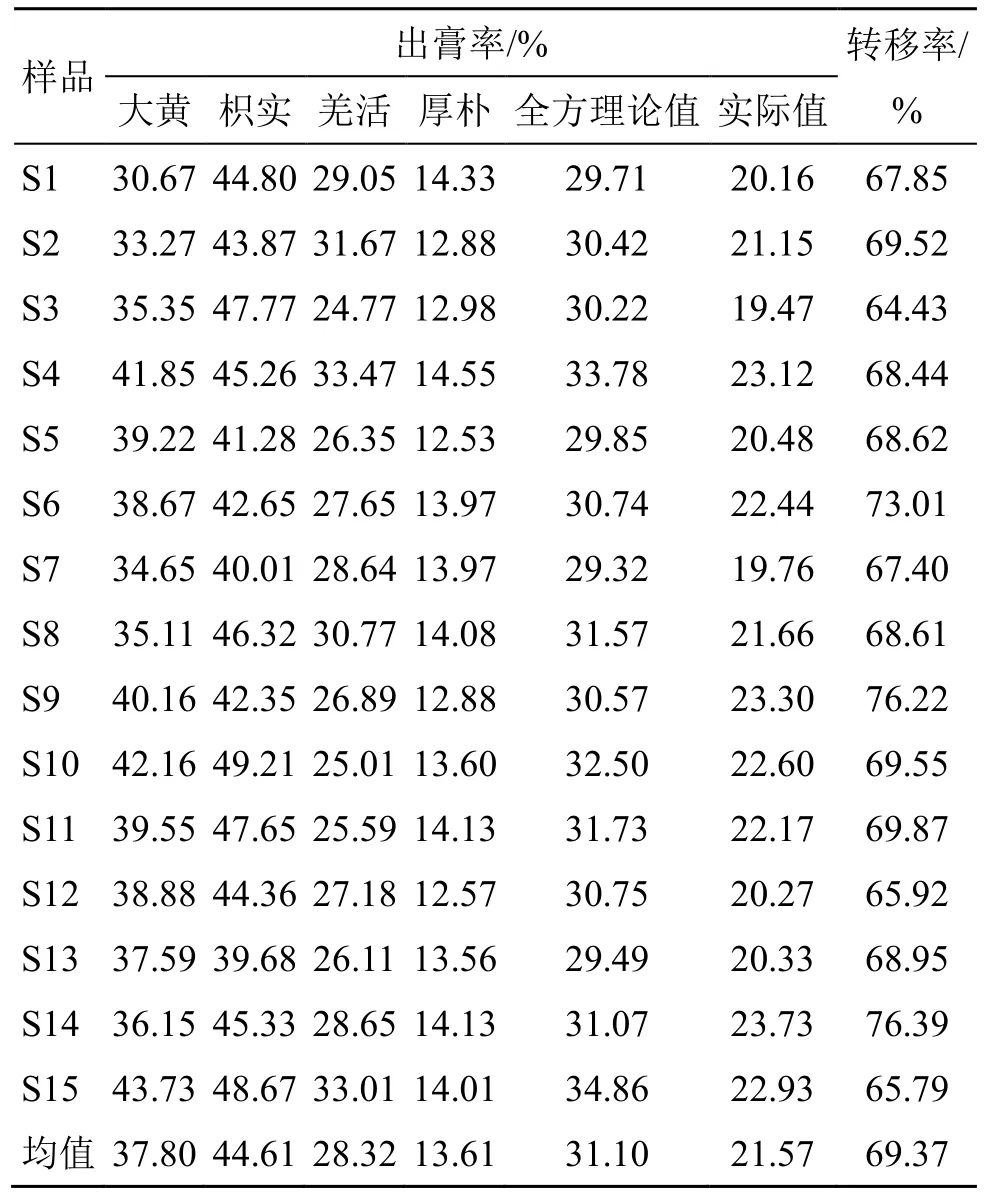
表8 三化汤基准样品出膏率及转移率Table 8 Paste yield rate and transfer rate of Sanhua Decoction benchmark samples
3 讨论
3.1 处方组成及计量考证
经典名方疗效确切且应用历史悠久,但存在着古今剂量换算及炮制差异等众多难点问题。经文献考证,金元与唐宋时期处方剂量折算类似[9-10],宋代1 两合今39~41 g[11-12],与国家中医药管理局和国家药品监督管理局公布的“古代经典名方关键信息表(25 首方剂)”信息中金时期1 两约为41.30 g 基本吻合,“每服三两”也就是每服处方117.00~123.00 g,“大黄、枳实、羌活、厚朴各等分”则四味药各29.25~30.75 g;参考苑祯等[13]对宋元时期“升”的考证,得到处方现代计量单位换算:金元时期1 升折合今约为702 mL。最终确定处方为大黄、枳实、羌活、厚朴均为生品各30 g,加水“三升”(2100 mL),煎至“一升半”(1050 mL)。
3.2 色谱条件分析
本实验采用UPLC 建立三化汤特征图谱和指标成分含量测定方法,前期实验分别采用乙腈-0.1%磷酸水溶液、甲醇-0.1%甲酸水溶液和乙腈-0.1%甲酸水溶液做流动相,经对比发现,采用乙腈-0.1%甲酸水溶液进行梯度洗脱时,基线平稳,色谱峰分离效果较好。
另外考察不同体积流量(0.2、0.3、0.4 mL/min)对色谱峰的影响,当体积流量升高时,主要成分色谱峰分离度较低,当体积流量控制在0.2 mL/min 时,色谱峰峰形较好。波长在250~260 nm 时,特征图谱出峰较多,整体成分信息较为全面;255 nm 波长下基线平稳,各成分色谱峰分离效果较好,故最终选择255 nm 作为特征图谱的最佳波长。
3.3 指标成分的选择及量值传递研究
基准样品中柚皮苷和新橙皮苷专属性强且含量远高于其他成分,在特征图谱中较为明显,2 成分在热水中水溶性较好,有研究证明,新橙皮苷对柚皮苷具有一定的增溶作用[14],且两者转移率均大于40%,前期实验研究发现辛弗林在含量测定图谱中有明显阴性干扰,因此未选择其作为枳实的测定指标成分。基准样品中紫花前胡苷相比于《中国药典》2020 年版规定的羌活测定指标成分异欧前胡素和羌活醇UPLC 分离效果更好,而且在进行TLC 鉴别过程中紫花前胡苷的分离度较好、表征明显,更有利于质量标准的建立,因此选择其作为指标成分之一。基准样品中游离总蒽醌、和厚朴酚与厚朴酚极性较低,作者采用三氯甲烷作提取溶剂制备基准样品中游离总蒽醌、和厚朴酚与厚朴酚含量测定供试品,有效降低了大极性成分对于检测指标的干扰,同时缩短了检测时间,并通过切换不同波长,增强了检测指标色谱峰信号。
以指标成分转移率在均值的±30%为衡量标准,15 批三化汤基准样品中紫花前胡苷、柚皮苷和新橙皮苷转移率结果均符合该规定,但部分批次和厚朴酚与厚朴酚总量、游离总蒽醌的转移率未在均值的±30%范围内,其中220105、220502 批次厚朴中明显略高于均值的30%,分析原因可能是制备过程中受煎煮环境等因素影响导致指标成分转移率相差较大,后期制剂生产可考虑适当剔除部分批次转移率过低的饮片或采用混批投料方式来保证量值传递的均一性。大黄中游离蒽醌类化合物与厚朴中厚朴酚已被证明均是三化汤发挥治疗脑卒中的重要成分[15-16],厚朴酚与和厚朴酚是同一化学结构的同分异构体,具有疏水性烯丙基联苯酚类结构[17],水溶性较差,转移率较低;随着提取时间增加,部分游离蒽醌并不稳定,会产生氧化分解[18],后期制剂生产中应改进提取工艺,以期提高药效成分含量。
经典名方基准样品作为制剂开发的中间体,是制剂前期生产的重要部分[19],更是建立饮片-中间体-制剂质量控制体系的关键,阐明方中关键成分量值传递规律,可以保证基准样品的均一、稳定[20-22]。本研究通过建立不同批次基准样品特征图谱对三化汤整体成分信息进行表征和相似度评价,并对其共有峰进行了指认与归属,所建立的特征图谱相似度均大于0.98,含量测定结果显示基准样品各指标成分转移率基本控制在均值的±30%,各批次处方干膏转移率均在均值的90%~110%,波动范围在规定的均值±10%,表明基准样品制备工艺及处方转移率较为稳定,可为后续三化汤制剂的进一步开发提供参考。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
现代临床医学(2021年1期)2021-01-26
通化师范学院学报(2020年12期)2020-12-21
海峡姐妹(2019年8期)2019-09-03
天然产物研究与开发(2019年1期)2019-03-01
天然产物研究与开发(2018年1期)2018-02-02
中成药(2017年12期)2018-01-19
中成药(2017年6期)2017-06-13
益寿宝典(2017年2期)2017-02-26
吉林大学学报(医学版)(2015年3期)2015-12-17
中国当代医药(2015年24期)2015-03-01
