
钯铋催化剂在乙炔加氢中的构效关系研究
2023-05-31 06:14:12李玉楠郭玲玲黄宇营李丽娜
核技术 2023年5期
陶 鑫 赵 贵 李玉楠 田 晨 杜 萌 郭玲玲 南 兵 黄宇营 李丽娜
1(中国科学院上海应用物理研究所 上海 201800)
2(中国科学院大学 北京 100049)
3(中国科学院上海高等研究院 上海同步辐射光源 上海 200120)
4(上海交通大学 物理科学学院原位电镜中心 上海 200240)
乙烯作为一种基本的石油化工产品需求量巨大,全球每年消耗约1.97亿吨。生产乙烯的传统方法是石油蒸汽裂解产生,但是需要高温和很高的生产成本[1-2]。因此,以电石(CaC2)路线为基础生产乙烯是一种很好的节约成本经济途径[3]。CaC2是一种常见的无机物,它能与水反应生成乙炔。乙炔半加氢制乙烯是一种经济有效的乙烯生产方法。
正如Teschner等[4]报道的那样,钯(Pd)催化剂的选择性差是因为乙烯加氢的势能比乙烯脱附的势能要小。此外,在乙炔加氢过程中容易形成氢化钯(PdHx)[5],表面乙烯很容易被进一步氢化为乙烷[6]。理论上,如果减弱乙烯的吸附和PdHx的形成就可以提高乙炔加氢的选择性。为了提高负载型Pd催化剂在乙炔加氢反应中的选择性,利用一氧化碳等分子作为修饰剂[7],与乙烯竞争吸附位点,有利于乙烯的解吸提高选择性。然而,CO的存在降低了氢的表面浓度,促进了绿油的生成和催化剂的失活。另外的方法是引入第二种金属来修饰钯催化剂的电子结构和几何结构。例如:Ag在Pd催化剂中可以减少氢的吸附量,抑制氢从本体向表面的扩散,减少氢化物的产生,从而提高乙烯的选择性[8]。Cu在Pd催化剂中减少了多配位Pd位点的数量,而多配位Pd位点是乙烷生成和乙烯选择性低的原因[9]。这类金属具有较高的烯烃选择性,但通常需要在高温下活化使用,这限制了广泛的工业使用[10-11]。
引入第二种金属修饰在热催化中有着广泛的应用[12]。 比 如 :析 氢 反 应(Hydrogen Evolution Reaction,HER)[13]、氧 化 还 原 反 应(Oxygen Reduction Reaction,ORR)[14]、氢化反应[9]、CO 氧化[15]等。修饰过后的催化剂能够提高贵金属的利用率,独特的电子结构与载体之间的强相互作用提高催化剂的活性与选择性。与Pd催化剂相比,Bi对于H原子的吸附较低[16],容易从表面脱附。我们用沉积沉淀法将Bi与Pd一起沉淀形成了Pd-Bi独特结构负载到SiO2的催化剂。出乎意料的是,在相同负载量下Pd-Bi结构催化剂表现出较高的活性和选择性。
1 材料与方法
1.1 试剂与仪器
本实验所用的所有化学试剂都是分析纯,并且在使用过程中没有进一步的纯化和修饰。反应气体购于液化空气(上海)压缩气体有限公司,直接使用未进行其他处理。
1.2 样品的制备
先将二氧化硅(SiO2)在450 ℃马弗炉中煅烧2 h,升温速率为5 ℃·min-1。在100 mL去离子水中加入1 g SiO2载体。用沉积沉淀法负载钯和铋,取一定量的硝酸钯(Pd(NO3)2·2H2O)作为前驱体,钯的负载量为1%(w/w,质量分数),溶解到10 mL的去离子水中。取一定量的硝酸铋(Bi(NO3)3·5H2O)作为前驱体溶液,前驱体的负载量为1%(w/w),溶解到10 mL的去离子水中。用0.1 mol·L-1的碳酸氢钠(NaHCO3)作为沉淀剂,逐滴加入,调节pH=7,70 ℃油浴3 h。将沉淀物离心,用去离子水洗涤5次,70 ℃过夜干燥,研磨将样品放在马弗炉450 ℃煅烧2 h,升温速率为5 ℃·min-1,得到所需催化剂,分别记为Pd/SiO2、PdBi/SiO2。
1.3 样品表征
1.3.1 ICP-AES实验
电感耦合等离子体发射光谱法(Inductively Coupled Plasma-Atomic Emission Spectrometry,ICP-AES)使用Optima 7300 DV仪器测定元素百分比。将样品溶于稀硝酸,80 ℃油浴回流,10 h后完全溶解,取一定量的溶液测Pd和Bi的含量。
1.3.2 粉末XRD实验
用 Bruker AXS D2 PHASER 在 Cu Kα(λ=0.154 06 nm)辐照下,用微米级氧化铝圆盘标定2θ角,测定了催化剂的衍射谱。研磨后,在每次测试前,催化剂被压平在石英玻璃样品夹上。衍射图谱采集范围为10°~90°,步长为0.08°。
1.3.3 氮气吸脱附实验
氮气吸附-脱附测量在77 K条件下在ASAP2020-HD88分析仪(Micromeritics Co)上进行,空气煅烧样品(新鲜催化剂)直接用于表征。粉末样品在真空下250 ℃脱气4 h后进行分析。相对压力(P/P0)在0.05~0.20范围内的数据Brunauer-Emmett-Teller(BET)进行计算。根据 Barrett-Joyner-Halenda(BJH)方法,从等温脱附曲线计算样品的孔径分布(Dp)。
1.3.4 透射电子显微镜
使用JEOL F200冷场发射透射电子显微镜和HITACHI HF5000球差校正扫描透射电子显微镜进行高角环形暗场扫描透射(High-Angle Annular Dark-Field Scanning Transmission Electron Microscopy,HAADF-STEM)和X射线能量散射光谱(Energy Dispersive Spectroscopy,EDS)表征,该仪器的工作电压为200 kV。测试前把微量的样品粉末加入无水乙醇,超声10 min,然后汲取一滴悬浊液滴到铜网上,该铜网包覆有超薄碳膜。滴有样品的铜网在放入电镜样品杆之前在红外灯下干燥10 min。
1.3.5 氢气程序升温还原实验
氢气程序升温还原(H2-TPR)和乙烯程序升温脱附(C2H4-TPD)在Micrometrics autochem 2920化学吸附仪用热导检测器(Thermal Conductivity Detector,TCD)检测。对于H2-TPR,将200 mg样品放在U形玻璃管中。用5% O2/He以10 ℃·min-1升温到300 ℃,保持0.5 h,去除表面水。降到室温,切换到5% H2/He以10 ℃·min-1升温到300 ℃进行H2-TPR实验,用TCD检测信号。
1.3.6 乙烯程序升温脱附实验
将200 mg样品放在U形玻璃管中,通入5%H2/Ar以 10 ℃·min-1升温到300 ℃并保持1 h还原催化剂。切换到氦气吹扫1 h,将温度降到-25 ℃后通入纯乙烯,在此温度下吸附1 h,切换到氦气以10 ℃·min-1进行乙烯程序升温实验,用TCD检测信号。
1.3.7 同步辐射XAFS表征
在上海同步辐射光源(Shanghai Synchrotron Radiation Facility,SSRF)14W线站进行X射线吸收精细结构(X-ray Absorption Fine Structure,XAFS)测试。使用Si(311)双晶体单色仪对X射线光束进行单色化处理。为了消除高次谐波,设置25%的失谐度。测试前,将粉末样品压成直径1 cm的薄片,用Lytle探测器荧光模式测试Pd K边和Bi L3边的XAFS光谱。用来对比的标样Pd foil、PdO、Bi foil和Bi2O3是在透射模式下测试的。所有采集的原始XAFS数据采用IFEFFIT软件包中的ATHENA模块进行处理,采用IFEFFIT软件包的ARTEMIS模块对K2加权的χ(k)函数进行傅里叶变换后在R空间进行定量曲线拟合[17-18]。
1.4 催化性能测试
将100 mg样品(40~60目)和200 mg石英砂(40~60目)混合装到石英管的中间。用5% H2/Ar混合气,以 10 ℃·min-1升温到 300 ℃还原催化剂 1 h。切换到Ar气吹扫到室温,通入1% C2H22% H2/Ar,以5 ℃·min-1升温到目标温度,每个温度点保持1 h空速 (Gas Hourly Space Velocity, GHSV) 为30 000 mL·h-1·gcat-1。 采 用 Agilent Technologies 7890B气相色谱配有火焰离子化监测器(Flame Ionization Detector,FID)来检测乙炔、乙烯、乙烷、1,3丁二烯的浓度。
C2H2转化率计算式为:
C2H4选择性计算式为:
2 结果与讨论
氮气吸脱附实验提供了载体的结构信息。吸附等温线为典型的IV型(图1)。当分压(P/P0)在0~1.0之间时,等温曲线也具有清晰的H1滞回线,这可能是由于介孔内部结构的毛细凝结和蒸发所致[19]。SiO2的 比 表 面 积 计 算 得 299.99 m2·g-1,孔 体 积0.77 cm3·g-1,孔径12.15 nm。SiO2比表面积大,催化剂的前驱体容易负载在SiO2上。
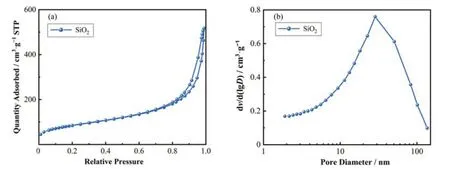
图1 N2吸脱附曲线(a)和载体SiO2的孔径分布(b)Fig.1 N2 adsorption-desorption isotherms (a) and pore size distributions of support SiO2 (b)
根据ICP-AES在表1中的结果,经过沉积沉淀法之后Pd的负载量都在1%(w/w)左右,说明Pd被沉淀到了SiO2上。Bi的负载量也接近目标负载量,Bi也是很好地负载到载体上了。
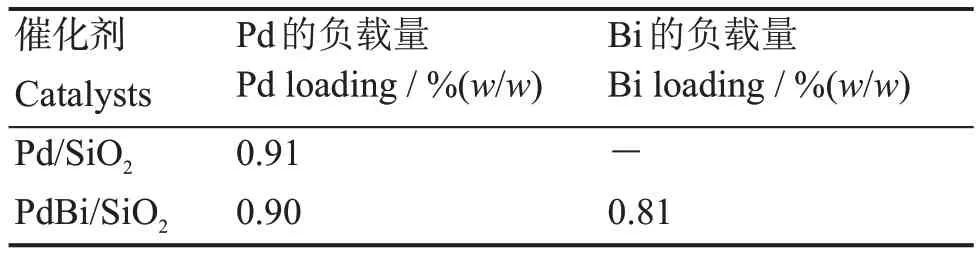
表1 PdBi/SiO2催化剂的元素组成Table 1 Elemental analysis of PdBi/SiO2 catalyst
采用X射线衍射(X-ray Diffraction,XRD)研究了掺杂不同Bi的催化剂晶体结构。XRD谱图给出了从2θ在10°~80°中峰强度和位置的分布,从图2可以看出,20.8°归属于SiO2的衍射峰,31.7°归属PdO的衍射峰。用Scherrer公式计算出PdO的颗粒大小为10.3 nm。从XRD图中可以看出,Pd/SiO2新鲜焙烧过后的样品有明显PdO的衍射峰。当加入Bi之后,PdO的衍射峰消失,说明了Bi的加入减小了PdO的尺寸或者使PdO的分散性变好。反应后的样品经过了300 ℃ H2的处理以及乙炔和氢气反应都没有出现Pd的衍射峰,Bi的引入使得Pd结构非常稳定。然而,Pd/SiO2反应过后的样品在39.1°出现了Pd单质的峰,反应后的样品Pd以单质形式析出来。在PdBi/SiO2整个谱图当中并没有发现Bi2O3的峰,Bi的颗粒可能太小而检测不到或者有极好的分散性。这个有趣的现象说明,Bi的加入有利于提高Pd的分散性,也有可能进入PdO的晶格里面形成Pd-O-Bi独特结构催化剂,Bi金属的引入极大修饰了PdO的尺寸。为了证明这个观点,我们采用环形暗场扫描透射电子显微镜(HAADF-STEM)研究了Pd在SiO2上的分散。从图3(a)可以看出,SiO2主要是无定形的,在SiO2上有小的亮点。图3(b)采用高倍电镜,可见在SiO2的表面上PdO分散性好,以大颗粒的形式存在SiO2的表面。由于PdBi/SiO2颗粒较小,用球差校正扫描透射电子显微镜进行高角环形暗场像表征PdBi/SiO2,从图 3(c、d)可以看出,PdBi/SiO2颗粒的大小为5.2 nm。我们对其中的一个颗粒做了线扫做元素分析发现,Pd和Bi之间的强度差不多可以证明颗粒是由PdBi两种元素形成的独特结构催化剂。

图3 HAADF-STEM图 (a, b) Pd/SiO2,(c, d) PdBi/SiO2,(e) PdBi/SiO2催化剂的EDS线扫结构Fig.3 HAADF-STEM images (a, b) Pd/SiO2, (c, d) PdBi/SiO2, (e) Linear scan EDS spectra of the catalyst PdBi/SiO2
为了进一步验证PdBi物种的结构,将PdBi/SiO2样品用5% H2/Ar在300 ℃处理1 h,然后在N2手套箱中进行压片处理,将样品记为PdBi/SiO2-Activation。我们用XAFS谱来研究PdBi/SiO2和Pd/SiO2以及4个标样Pd foil、PdO、Bi foil和Bi2O3。X射线吸收近边 结 构(X-ray Absorption Near Edge Structure,XANES)光谱表明,PdBi/SiO2和Pd/SiO2与PdO的近边接近说明Pd的价态是接近2价(图4(a))。采用Pd foil和PdO的标样,对相应的XANES进行线性组合拟合,确定其Pd/SiO2和PdBi/SiO2中Pd的平均氧化均为+2价(0% Pd和100% PdO)(图4(a、b))。采用Bi foil和Bi2O3的标样,对相应的XANES进行线性组合拟合,确定PdBi/SiO2中Bi的平均氧化态为+2.7 价(8.7% Bi和 91.3% Bi2O3)(图 4(c))。PdBi/SiO2-Activation中Bi的平均价态为+0.6价(77.5% Bi和22.5% Bi2O3)(图4(d))。如图5所示,傅里叶变换(FT)K2加权扩展X射线吸收精细结构(EXAFS)提供了样品的配位结构。表2中,Pd/SiO2中R=2.02 Å,CN=4,PdBi/SiO2中,R=2.01 Å,CN=3.5。Bi的加入对第一壳层Pd-O无影响,而Pd/SiO2在第二壳层Pd-O-Pd中CN=10。在PdBi/SiO2第二壳层拟合出Pd-O-Bi而不是Pd-O-Pd,其中Pd-O-Bi的配位数CN=1.7。Bi的引入降低了第二壳层的配位数,进入了PdO里面形成了Pd-O-Bi结构。低配位数的Pd可以提高乙炔加氢的选择性[9]。为了探究Bi在上面影响,拟合了Bi的第一壳层,Bi-O中CN=4,没有Bi-OBi壳层,Bi在Pd表面上是以单原子的形式存在的。PdBi/SiO2-Activation中形成了Bi-Pd键,其中R=2.79 Å,CN=3.5。证明了在经过300 ℃的H2处理后,Pd与Bi之间形成了Bi-Pd独特结构的催化剂。
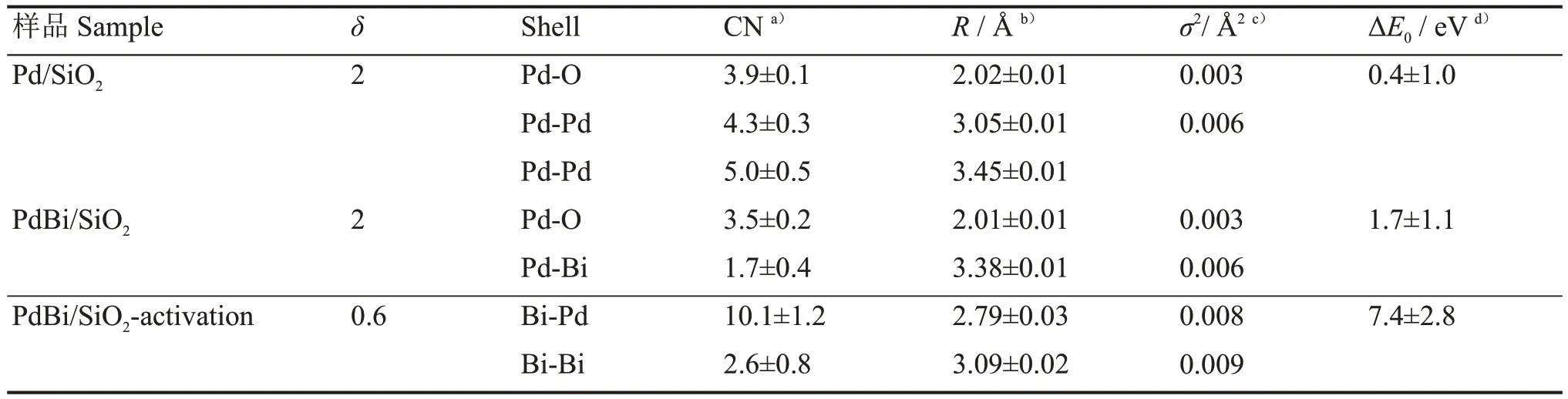
表2 钯和铋价态(δ)PdBi/SiO2中Pd K边和Bi的L3边EXAFS拟合数据Table 2 Oxidation state of palladium and bismuth (δ) Curvefit parameters for Pd K-edge and Bi L3-edge EXAFS for PdBi/SiO2
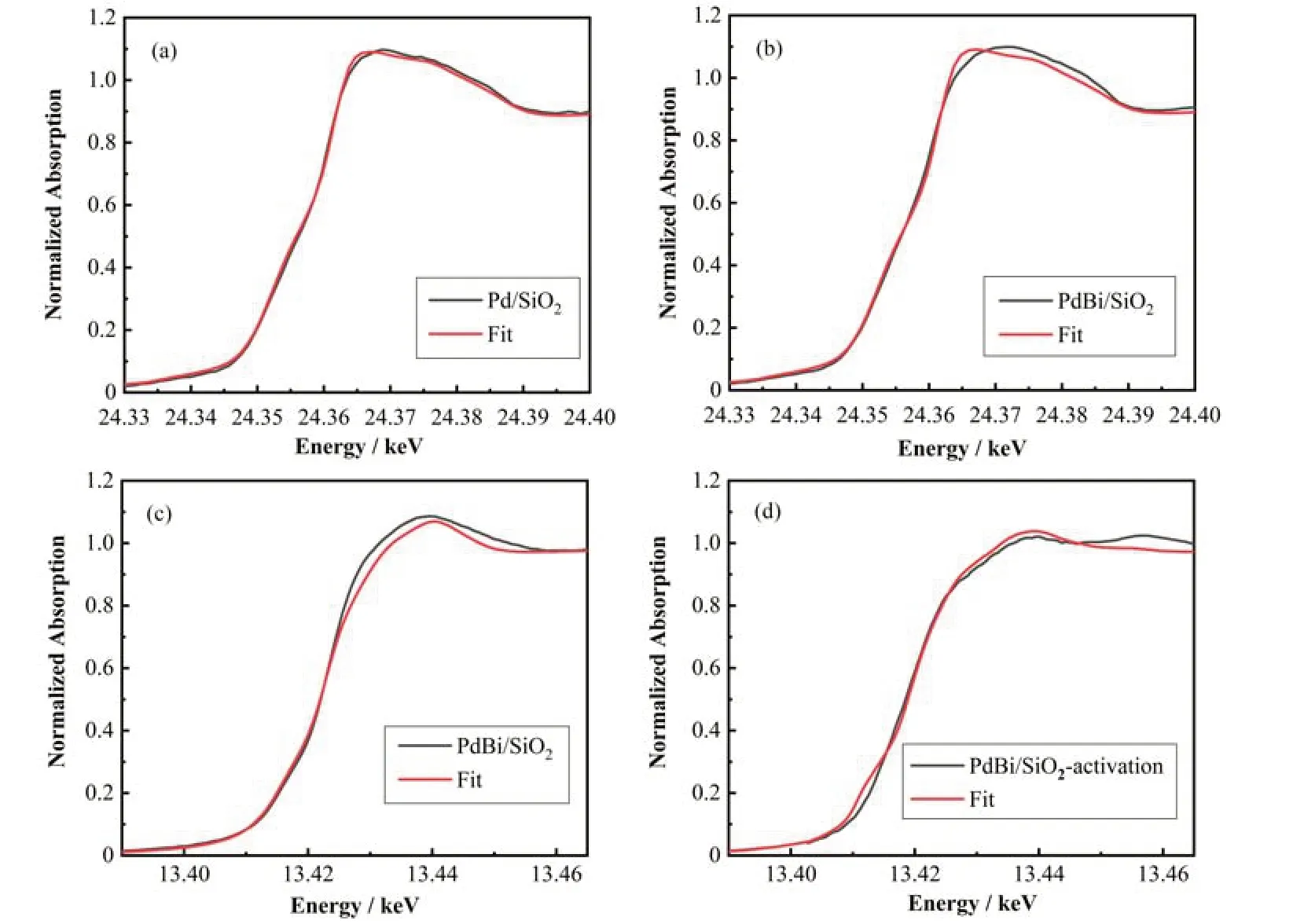
图4 Pd/SiO2样品中PdK边的近边拟合 (a) Pd/SiO2,(b) PdBi/SiO2、PdBi/SiO2中Bi L3边的近边拟合,(c) PdBi/SiO2,(d) PdBi/SiO2活化后样品Fig.4 The linear combination fitting reasults of Pd K edge of catalysts (a) Pd/SiO2, (b) PdBi/SiO2, the linear combination fitting reasults of Bi L3 edge of catalysts, (c) PdBi/SiO2 , (d) PdBi/SiO2-activation
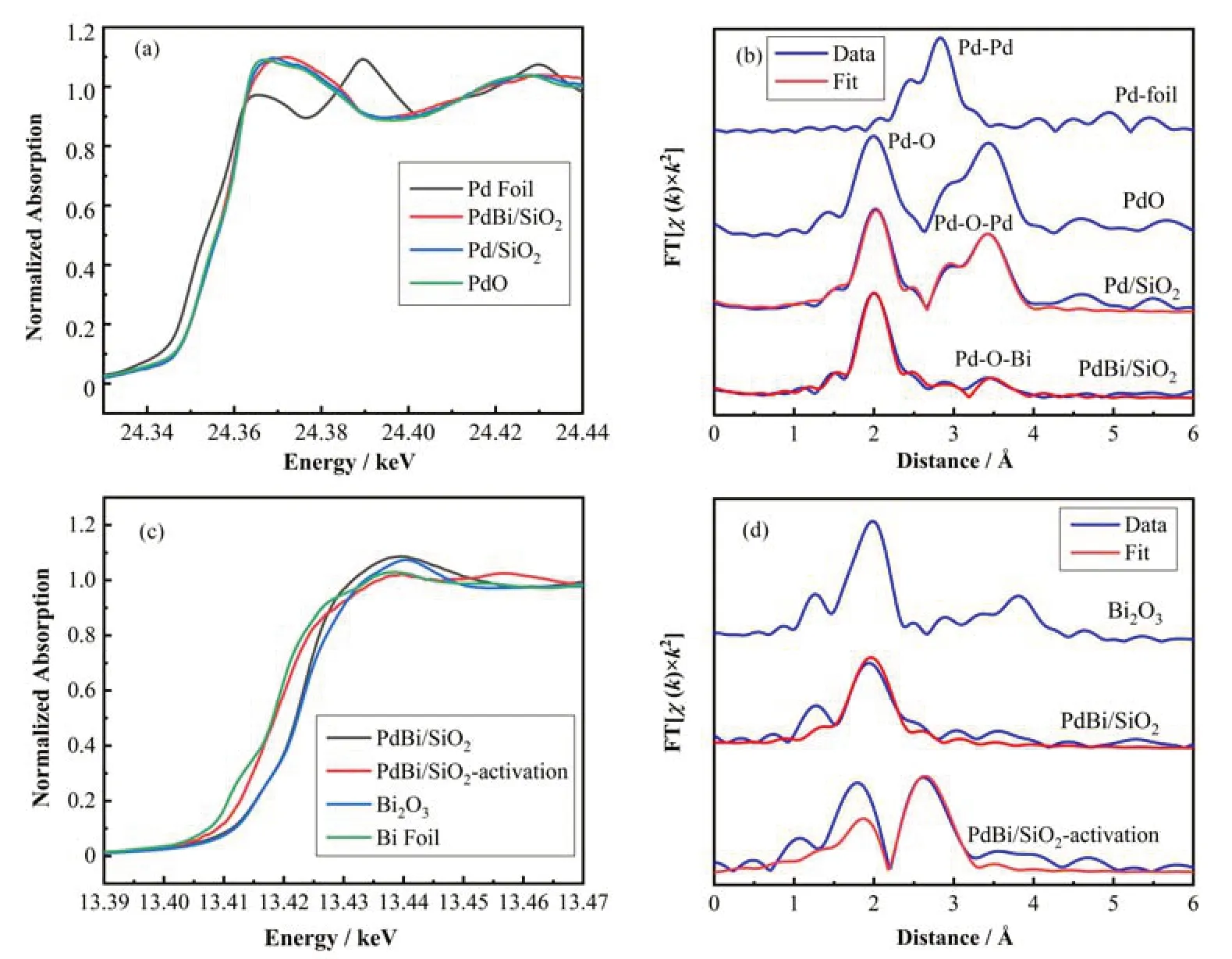
图5 Pd/SiO2样品中Pd K边近边吸收(a)与扩展边结构(b),PdBi/SiO2样品中Bi的L3边近边吸收(c)与扩展边结构(d)Fig.5 XANES profiles (a) and EXAFS spectra (b) of the Pd/SiO2 catalysts, XANES profiles of Bi L3 in PdBi/SiO2 (c) and EXAFS spectra of Bi (d)
对催化剂进行乙炔半加氢性能测试,结果如图6所示。Pd/SiO2在30 ℃下转化率达到80%,选择性却不到60%。当温度提高到50 ℃时,乙炔的转化率达到了100%,实现了完全转化,乙烯的选择性却只有36.4%。随着温度的提高,乙炔的转化率不变,乙烯选择性逐渐降低。PdBi/SiO2催化剂温度达到50 ℃时,乙炔的转化率降低到50%,而乙烯的选择性提高到93.4%。随着温度的提高,乙炔转化率逐渐提高而乙烯选择性缓慢降低。我们要探究在乙炔完全转化时乙烯的选择性,在100 ℃的时候,PdBi/SiO2实现了完全转化且选择性在90%以上,说明了Bi的加入虽然降低了转化率但是提高了选择性,避免了乙炔过度加氢生成乙烷。为了进一步说明Bi的加入对催化剂还原温度的影响,H2-TPR实验中SiO2上没任何峰,说明载体是惰性材料没有还原性。Pd/SiO2在48.9 ℃左右的氢气吸收是由于发生了PdO到PdHx的还原,这是因为在Pd的表面上形成了氢溢流现象[20]。我们发现加入了Bi之后,PdBi/SiO2在100 ℃只有PdO的峰没有氢溢流现象,氢溢流现象的消失是选择性提高的原因。C2H4-TPD是为了探究C2H4在催化剂表面的脱附能力,Pd/SiO2和PdBi/SiO2乙烯脱附的温度分别为77.8 ℃和88.9 ℃,证明了Bi的加入降低了乙烯的脱附温度。在乙炔加氢生成乙烯过程中,PdBi/SiO2表面生成了乙烯之后更容易脱附掉,避免了乙烯吸附于表面加氢生成乙烷降低了选择性。

图6 催化剂性能评价 (a) Pd/SiO2和PdBi/SiO2乙炔转化率随着温度的变化,(b) Pd/SiO2和PdBi/SiO2乙烯选择性随温度的变化,(c) H2-TPR,(d) C2H4-TPDFig.6 Catalyst perform (a) C2H2 conversion and (b) C2H4 selectivity as a function of temperature over the different catalysts, (c) H2-TPR, (d) C2H4-TPD
3 结语
利用沉积沉淀法成功将Pd和Bi沉淀到SiO2的表面。PdBi/SiO2具有更高的乙炔选择性加氢活性和乙烯选择性。PdBi独特结构催化剂的形成极大提高了贵金属的原子利用率。由于Bi对H原子的吸附差,H2在Pd表面裂解形成H原子溢流到Bi的表面脱附出去避免了PdHx的形成,C2H4在催化剂表面避免过度加氢形成C2H6,提高了催化剂的选择性。这些实验结果为Bi对Pd促进工业应用创造了更多的潜力。
致谢感谢上海光源BL11B线站老师在光路优化和XAFS实验过程中给予的帮助,感谢上海光源用户辅助实验组给予实验的支持,感谢上海交通大学原位电镜中心在球差电镜的实验支持。
作者贡献说明陶鑫负责研究的提出及设计、数据的收集和整理、文章的起草和最终版本的修订;赵贵负责电镜的拍摄;李玉楠负责样品的合成;田晨负责吸收谱的表征;杜萌负责活性测试;郭玲玲、南兵负责数据解析;黄宇营、李丽娜负责最终版本的修订。
猜你喜欢
化学工程师(2023年1期)2023-02-17 15:09:48
理化检验-化学分册(2020年12期)2021-01-26 00:41:38
中国果树(2020年2期)2020-07-25 02:14:28
上海农业科技(2019年1期)2019-02-22 01:51:28
科普创作(2018年1期)2018-11-30 05:11:40
石油化工建设(2018年2期)2018-07-11 01:25:04
电源技术(2016年9期)2016-02-27 09:05:25
自动化博览(2014年9期)2014-02-28 22:33:35
自动化博览(2014年8期)2014-02-28 22:33:06
无机化学学报(2014年9期)2014-02-28 17:33:05
