
利用Cas9/sgRNA体系提高猪胎儿成纤维细胞基因组编辑效率
2023-05-29 03:01:32姜爱文郭鸿运吴望军刘红林
南京农业大学学报 2023年3期
姜爱文,郭鸿运,吴望军,刘红林
(南京农业大学动物科技学院,江苏 南京 210095)
我国是世界上最大的猪肉消费国,猪(Susscrofa)作为我国重要的经济动物,其繁殖性状、猪肉产量和肉质性状的提高始终是猪遗传育种工作的主要方向[1-2]。传统的猪育种技术依托于纯系选育和配套杂交等方法,对生长速度、饲料转化率等遗传性状的改良,往往需要多代杂交和选择才能获得优质个体[3]。传统育种所需时间长,无法突破物种界限引入其他新型性状,且其在瘦肉率、肌内脂肪含量(intramuscular fat,IMF)、背膘厚等数量性状的选择上准确度低、选育费用高。在快速发展的当今社会,传统育种方法已无法满足人们日益增长的消费需求。分子育种技术,如全基因组关联分析(genome wide association study,GWAS)的发展,可将基因型的多样性与表型性状相结合,以获得影响复杂性状的分子遗传标记和候选基因。Fan等[4]利用GWAS发现黑素皮质素受体4(melanocortinc 4 receptor,MC4R)是影响背膘厚的候选基因;Luo等[5]鉴定到了与肉质性状有关的47个单核苷酸多态性(single nucleotide polymorphisms,SNP)位点;韩云珍等[6]发现MC4R基因的多态性与美系大白猪的胴体性状密切相关,这些研究为加速常规育种进程提供了可借鉴的信息。近年来,随着基因编辑技术的兴起,人为干预基因组信息,并对目的基因进行插入或删除,可以直接、迅速培育出特定性状的家畜,也可以打破生殖隔离,实现不同物种之间的交流。包括转基因、基因编辑在内的“分子设计育种”已成为分子育种的重要研究方向。此外,由于体型大小和特征与人类相似,猪也被认为是器官移植和人类疾病的理想模型[1,7-8]。因此,猪基因编辑技术的研究与应用不仅能加快家畜遗传育种的进程,也对人类再生医学具有重要意义。
基因组编辑技术主要包括位点定向锌指核酸酶(zinc finger nucleases,ZFN)、转录激活因子样效应核酸酶(transcription activatorclike effector nucleases,TALEN)和聚类规则间隔短回文重复序列/相关核酸酶Cas9(clustered regularlyinterspaced short palindromic repeats,CRISPR/Cas9)3种[9-11]。其中,CRISPR/Cas9系统由于具有成本低、操作简单、效率高等优点,已成为目前最常用的基因编辑方法[12]。基因组编辑技术的发展加速了动物育种的进程。与传统的杂交方法不同,基因组编辑可以在小鼠、人、羊、猪等多种物种中快速有效修改目的基因序列[13]。目前,基因编辑猪在抗病性和提高肉质等方面取得了很大进展。肌肉生长抑制素(myostatin,MSTN)是骨骼肌生长发育的负调控因子,其失活可诱导骨骼肌肥大并促进肉类生产[14-15]。与野生型猪相比,MSTN基因敲除猪具有更高的肌生成素表达和明显的肌间边界,能显著促进猪的生长,增加瘦肉产量[16-17]。清道夫受体(cluster of differentiation,CD163)是猪繁殖与呼吸综合征(porcine reproductive and respiratory syndrome,PRRS)病毒的受体,CD163基因的敲除可以有效阻止猪感染PRRS病毒。Tanihara等[18]利用CRISPR/Cas9技术成功构建了CD163基因敲除猪,获得了PRRS病毒耐受模型。
基因编辑在应用转化中的最大阻碍源于对其安全性的担忧,如何保证外源基因高效插入并表达,而又不会影响基因组本身的功能成为应用基因敲入技术的基础。ROSA26是一种非编码RNA。在小鼠中,ROSA26位于第6号染色体,最早在ROSA β geo26的小鼠品系中发现[19]。在后续研究中,研究人员发现ROSA26位点随机插入LacZ报告基因后,在小鼠的几乎所有组织中均能检测到β-半乳糖苷酶的高表达,且外源基因插入的小鼠生长与繁殖性能均无异常,ROSA26位点附近的基因表达也未受影响[20]。因此,ROSA26被认为是外源基因插入的“安全位点”,可以使外源片段安全、高效插入基因组。除了诱导外源基因的广泛表达外,ROSA26位点还可以结合Cre-LoxP条件敲入系统,诱导外源基因在特定组织或特定时间表达[21-22]。2014年,Li等[23]通过多个物种间比较,确定了猪ROSA26启动子区的序列,并利用TALEN技术成功地在猪ROSA26位点定点插入了Cre重组酶报告基因。随着CRISPR/Cas9技术在猪上的应用,猪防御素2(porcine β defensing 2,PBD2)、干扰素诱导基因S-腺苷甲硫氨酸基区域蛋白2(sadicalS-adenosyl methionine domain containing 2,RSAD2)等转基因猪相继获得[24-25]。
虽然基因编辑猪已成功获得,但低敲除效率阻碍了转基因动物产生过程中外源基因的敲入效率,并降低了体细胞核转移(somatic cell nuclear transfer,SCNT)的应用。因此,建立一个稳定、高效的基因敲除系统对促进基因编辑在猪上的应用是必要的。
1 材料与方法
1.1 试验材料
猪胎儿成纤维细胞(PEF)由本课题组分离、培养、建立。PX458-cas9-egfp质粒和PX459-cas9-puro质粒由安徽农业大学于童赠送;2个质粒均为cas9和sgRNA二合一质粒,其中PX458-cas9-egfp质粒可表达绿色荧光,在本研究中用于PEF电转体系的筛选;PX459-cas9-puro用于构建猪ROSA26的sgRNA,以实现对PEF基因组编辑。RGS-CR质粒由广西大学唐小川赠送。Cas9蛋白(A50576)和NeonTM转染试剂盒(MPK10025)购自ThermoFisher Scientific。与Cas9蛋白在体外形成RNP的sgRNA序列为sgRNA2序列(不含PAM),序列由ThermoFisher Scientific合成并修饰后使用。
1.2 试验方法
1.2.1 猪ROSA26打靶位点的选择根据猪ROSA26的基因组序列,在其启动子区域(含第一外显子)设计2条sgRNA(sgRNA1和sgRNA2)。sgRNA1/sgRNA2的具体序列为5′-AAAGTGTGCTGTGTATTTTG-3′/5′-AGAGAAGAGGCTGTGCTCTG-3′。
1.2.2 PX459-cas9-sgRNA打靶载体的构建根据sgRNA1和sgRNA2的序列,两侧添加BbsⅠ的黏性末端,合成sgRNA上游引物和下游引物(表1);上、下游引物磷酸化(NEB,M0201S)与退火后(NEB,B0202S)形成带有BbsⅠ黏性末端的短DNA双链。限制性内切酶BbsⅠ(NEB,R0539S)切割PX459-cas9-puro载体,再利用胶回收试剂盒(OMEGA,D2500-02)回收PX459-cas9-puro大片段。将sgRNA退火形成的短DNA双链与回收的PX459-cas9-puro大片段使用T4连接酶(NEB,M0202S)连接,随后经感受态细胞DH5α转化后进行菌液测序。对测序正确的菌液提取质粒(TIANGEN,DP117)。

表1 本试验所用引物序列Table 1 Sequence of primers used in this test
1.2.3 RGS-sgRNA载体的构建RGS-CR由pegfp-N1改造而成,红色荧光蛋白(red fluorescent protein,RFP)和增强型绿色荧光蛋白(enhanced green fluorescent protein,EGFP)由CMV启动子启动表达。在egfp序列前引入EcoRⅠ和BamHⅠ限制性内切酶的酶切位点,可使sgRNA靶序列在2个酶切位点间插入。在egfp前引入终止密码子,因此当没有Cas9/sgRNA切割靶位点时,EGFP不表达;当Cas9/sgRNA切割靶位点时,终止密码子发生移码突变,可使egfp有2/3概率处于开放阅读框内而发生表达。根据2条sgRNA的序列,在RGS-sgRNA上游引物中引入EcoR Ⅰ的酶切位点,在RGS-sgRNA下游引物中引入BamHⅠ的酶切位点,分别合成RGS-sgRNA的上、下游引物(表1)。上、下游引物退火形成短DNA双链;RGS-CR骨架序列用EcoRⅠ(NEB,R3101S)和BamHⅠ(NEB,R3136S)酶切后回收,使用T4连接酶连接,随后经感受态细胞DH5α转化后进行菌液测序。对测序正确的菌液进行质粒提取。
1.2.4 细胞培养与转染PEF细胞在含15%胎牛血清(Gibco,16000-044)和双抗(Gibco,15140-122)的DMEM培养基(Hyclnoe,SH30022.01B)中培养,待细胞融合度达到80%左右时,用PBS(Hyclone,SH30256.01B)洗2次,胰酶(Gibco,25200-072)消化3 min,随后加入15%培养基终止消化。消化后的细胞转移至15 mL离心管(Corning,430791)中,离心后获得细胞沉淀;将细胞沉淀用PBS重悬后离心,获得细胞沉淀。在24孔板上,细胞沉淀用5 μL Buffer R重悬,使细胞密度达到1.0×107mL-1;用5 μL Buffer R分别稀释 1 μg PX458-cas9-egfp质粒和1 μg PX459-cas9-puro质粒或Cas9/sgRNA2复合物(1 250 ng Cas9+240 ng sgRNA2);将含有细胞的Buffer R与含有质粒或蛋白的Buffer R混匀,使用Neon电转系统(MPK5000,Invitrogen)进行电转。电转3 d后,收集细胞,提取基因组DNA后进行打靶验证。取293T细胞在含10%胎牛血清和双抗的培养基中培养,待细胞融合度至80%左右,使用Lip3000(InvitrogenTM,L3000015)进行转染。转染3 d后在荧光倒置相差显微镜(Olympus,CKX41)下观察荧光。绿色荧光的激发光为488 nm,红色荧光的激发光为567 nm。
1.2.5 打靶效率检测PX459-cas9-sgRNA2质粒或Cas9/sgRNA2核糖核蛋白复合体转染3 d后,PEF细胞经PBS洗1次后收集细胞沉淀。使用基因组DNA提取试剂盒(TIANGEN,DP304)提取PEF细胞的基因组DNA。将PX459-cas9-sgRNA2质粒或Cas9/sgRNA2打靶后的基因组DNA浓度统一至30 ng·μL-1。基因组DNA使用Q5高保真酶(NEB,S0494)对打靶区域进行扩增。扩增引物为Target-F/R(表1),扩增片段的大小为360 bp。扩增体系:Q5高保真酶12.5 μL,10 μmol·L-1Target-F/R各1.25 μL,基因组DNA 1 μL,无DNA核酸酶水9 μL。混匀上述试剂,再按照如下程序进行PCR扩增:98 ℃ 30 s;98 ℃ 50 s,68 ℃ 10 s,72 ℃ 20 s,35个循环;72 ℃ 2 min;4 ℃保存。PCR产物经琼脂糖凝胶电泳鉴定,确定条带位置正确后进行TA克隆连接与测序。
1.2.6 脱靶位点鉴定sgRNA2潜在的脱靶位点(评分>1.5)见表2。为了检测sgRNA2的脱靶效应,经PX459-cas9-sgRNA2切割后的PEF细胞被收集并分别用引物进行扩增。引物的序列见表3。扩增后PCR产物进行桑格尔测序分析。
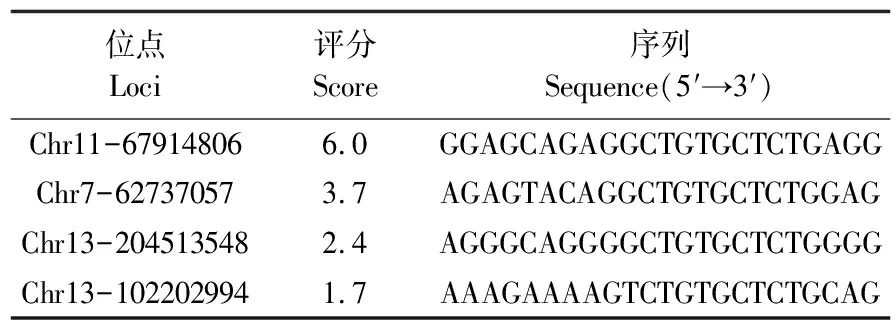
表2 脱靶位点信息Table 2 Information of off-target loci

表3 脱靶引物Table 3 Off-target primers
1.2.7 测序分析桑格尔测序分析由北京擎科生物科技有限公司完成。PX459-cas9-sgRNA质粒鉴定使用测序引物CAG-R(表1),RGS-sgRNA质粒鉴定使用测序引物CMV-F(表1),基因组DNA打靶检测的测序引物为Target-F/R(表1),脱靶检测采用正向引物进行测序(表3)。测序结果经DNAMAN比对分析。
1.3 数据分析

图1 PX459-cas9-sgRNA1和PX459-cas9-sgRNA2质粒的构建及测序验证Fig.1 Construction and sequencing validation of PX459-cas9-sgRNA1 and PX459-cas9-sgRNA2 plasmidsa、b.分别把sgRNA1和sgRNA2的上、下游引物退火后构建在PX459-cas9-puro质粒上,获得PX459-cas9-sgRNA1和PX459-cas9-sgRNA2质粒 The forward and reverse primer of sgRNA1 and sgRNA2 were annealed and then constructed into PX459-cas9-puro to obtain PX459-cas9-sgRNA1 and PX459-cas9-sgRNA2 plasmids;c、d.分别对PX459-cas9-sgRNA1和PX459-cas9-sgRNA2质粒进行桑格尔测序鉴定sgRNA1和sgRNA2插入片段 Identification of sgRNA1 and sgRNA2 sequences in PX459-cas9-sgRNA1 and PX459-cas9-sgRNA2 plasmids by Sanger sequencing.
2 结果与分析
2.1 PX459-cas9-sgRNA质粒的构建和验证
针对猪ROSA26位点的启动子区(含第一外显子)设计sgRNA1和sgRNA2,再将sgRNA序列插入PX459-cas9-puro质粒。sgRNA1和sgRNA2的质粒图谱见图1-a和图1-b。桑格尔测序结果表明,sgRNA1和sgRNA2均成功插入PX459质粒,分别获得PX459-cas9-sgRNA1和 PX459-cas9-sgRNA2(图1-c,d)。
2.2 RGS-sgRNA质粒的构建和验证
将sgRNA1和sgRNA2的靶序列及其PAM序列插入RGS-CR质粒。sgRNA1和sgRNA2的质粒图谱见图2-a和图2-b。桑格尔测序结果表明,sgRNA1和sgRNA2的靶序列及其PAM序列插入成功,分别获得RGS-sgRNA1和RGS-sgRNA2质粒(图2-c,d)。

图2 质粒RGS-sgRNA1和RGS-sgRNA2的构建及测序验证Fig.2 Construction and sequencing validation of RGS-sgRNA1 and RGS-sgRNA2 plasmidsa、b.RGS-sgRNA1和RGS-sgRNA2的上、下游引物退火后构建在RGS-CR质粒上,获得RGS-sgRNA1和RGS-sgRNA2质粒The forward and reverse primer of RGS-sgRNA1 and RGS-sgRNA2 are annealed and then constructed into RGS-CR to obtain RGS-sgRNA1 and RGS-sgRNA2 plasmid;c、d. 对RGS-sgRNA1和RGS-sgRNA2质粒进行桑格尔测序鉴定sgRNA1和sgRNA2插入片段 Identification of sgRNA1 and sgRNA2 sequence in RGS-sgRNA1 and RGS-sgRNA2 plasmids by Sanger sequencing.
2.3 sgRNA打靶效率的筛选
为比较sgRNA1和sgRNA2的打靶效率,PX459-cas9-sgRNA1/RGS-sgRNA1及PX459-cas9-sgRNA2/RGS-sgRNA2分别共转染293T细胞,并以RGS-CR为对照。RGS-CR质粒本身发红色荧光,而被打靶后能同时发出红色荧光和绿色荧光。结果表明,3组处理均可发出红色荧光,表明RGS转染成功(图3-a),且细胞转染效率在3组之间无显著差异(图3-b)。RGS-CR组无绿色荧光,但PX459-cas9-sgRNA1/RGS-sgRNA1和PX459-cas9-sgRNA2/RGS-sgRNA2转染后均能发出绿色荧光(图3-a),这表明sgRNA1和sgRNA2都能对各自的靶序列进行打靶。但sgRNA1的打靶效率显著低于sgRNA2(3-c),表明sgRNA2具有更高的打靶效率。因此,后续使用sgRNA2对猪PEF基因组进行打靶。
2.4 转染条件的筛选
为获得较高的PEF转染效率,使用Neon电转系统,将PX458-cas9-egfp转染至PEF中,并对合适的电转条件进行探索(图4-a)。由于PX458-cas9-egfp转染成功后细胞内可检测到绿色荧光,通过计算EGFP阳性细胞占总细胞的比例计算各电转条件的电转效率。由图4结果可知:1 500 V、3 t、10 ms的电转条件具有较强的细胞存活率,且细胞生长状态良好;1 600与1 650 V的电压使细胞形态明显皱缩,存活细胞量显著降低(图4-a,b);此外,在1 500 V、3 t、10 ms的电转条件下有较高的电转效率(图4-a,c)。因此,后续采用1 500 V、3 t、10 ms的条件对PEF进行电转。
2.5 脱靶鉴定
收集经PX459-cas9-sgRNA2打靶后的PEF细胞,提取基因组DNA后进行PCR扩增,利用NCBI获得的猪基因组序列并进行桑格尔测序。结果(图5)显示sgRNA2未发生脱靶。
2.6 PX459-cas9-sgRNA2与Cas9/sgRNA2打靶效率的比较
采用1 500 V、3 t、10 ms的电转条件,分别将PX459-cas9-sgRNA2质粒和Cas9/sgRNA2体系电转至PEF细胞中,再收集的DNA经扩增鉴定后使用Target-F/R引物进行桑格尔测序。测序结果(图6)表明,PX459-cas9-sgRNA2的打靶效率为15.67%,而Cas9/sgRNA2的打靶效率为33.33%。

图3 sgRNA打靶效率的筛选Fig.3 Screening sgRNA with higher efficiencya.PX459-cas9-sgRNA与RGS-sgRNA质粒共转至293T细胞中(红色荧光代表转染后效果,绿色荧光代表切割后效果,标尺=1 000像素)Co-transfection of PX459-cas9-sgRNA with RGS-sgRNA into 293T cells(Transfection efficiency is indicated by red fluorescent protein RFP,and cutting efficiency is indicated by green fluorescent protein. Scar bar=1 000 pixel);b.各组的转染效率Transfection efficiency among each group;c.各组的打靶效率Targeting efficiency among each group.不同字母表示不同组别间存在显著差异(P<0.05)。Different letters indicate significant difference among groups(P<0.05).

图4 Neon系统电转染条件的筛选Fig.4 Screening transfection conditions for Neon transfection systema.不同转染条件下PX458-egfp质粒效果观察(标尺=500像素)Effect of PX458-egfp plasmid under different transfection conditions(Scar bar=500 pixel);b.不同转染条件下细胞存活率Cell survival rate under different transfection conditions;c.不同转染条件下的转染效率Transfection efficiency under different transfection conditions.
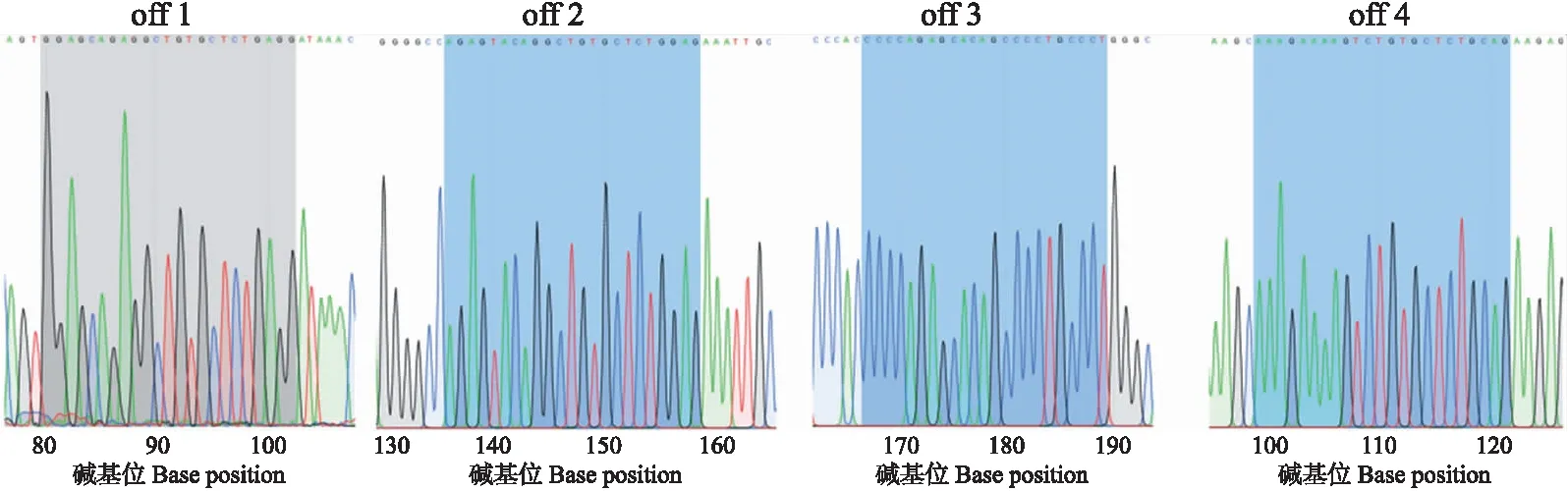
图5 sgRNA2排名前4位潜在脱靶位点的桑格尔测序检测Fig.5 Top 4 of potential off-target identification of sgRNA2 by Sanger sequencing
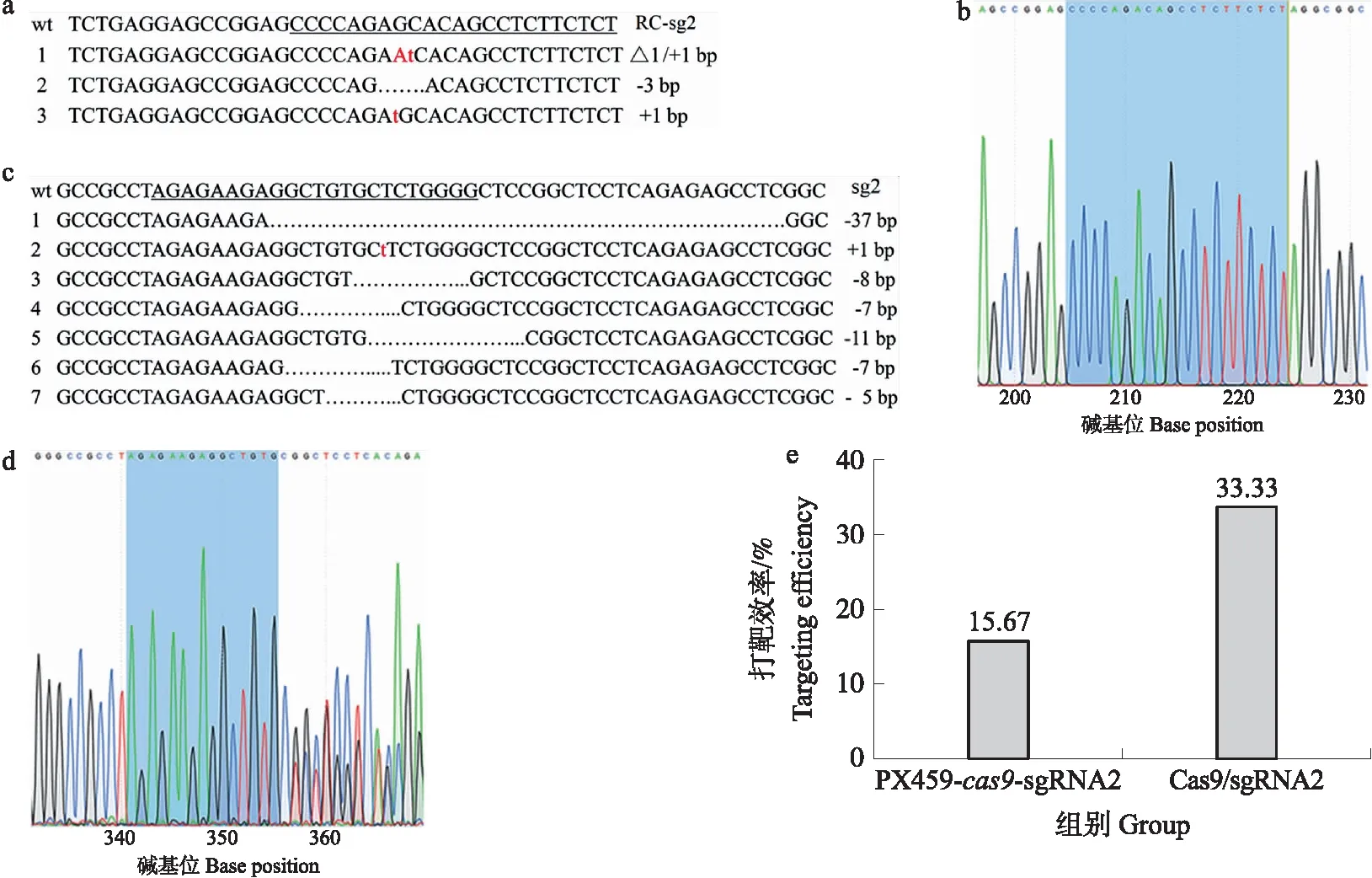
图6 PX459-cas9-sgRNA和Cas9/sgRNA系统切割打靶效率的比较Fig.6 Comparison of targeting efficiency between PX459-cas9-sgRNA plasmid and Cas9/sgRNAa.桑格尔测序检测PX459-cas9-sgRNA2质粒打靶猪胎儿成纤维细胞基因组的效率(第1行为野生型基因组序列,打靶后的序列位于第1行下面)The genome DNA sequence is used to analyze indels by Sanger sequencing in the group of PX459-cas9-sgRNA2 plasmid(The wild-type sequence is located on the first line(wt),and the mutated sequences are arranged below;b.PX459-cas9-sgRNA2打靶后的PEF基因组靶位点序列 Target sequence of PEF genome after PX459-cas9-sgRNA2 cleavage;c.桑格尔测序检测Cas9/sgRNA体系打靶猪胎儿成纤维细胞基因组的效率(第1行为野生型基因组序列,打靶后的序列位于第1行下面)The genome DNA sequence is used to analyze indels by Sanger sequencing in the group of Cas9/sgRNA(The wild-type sequence is located on the first line(wt),and the mutated sequences are arranged below;d.Cas9/sgRNA2体系打靶后的PEF基因组靶位点序列 Target sequence of PEF genome after Cas9/sgRNA2 cleavage;e.2组的打靶效率比较Cut efficiency between two groups.
3 讨论
随着CRISPR/Cas9技术的发展,基因编辑技术已经在多个动物物种上应用,包括小鼠、鸡、兔子、猪、羊[26-30]。目前基因编辑动物的获得主要有2种方式。一种是将cas9 mRNA和sgRNA共注射到胚胎干细胞中产生基因编辑动物。然而,猪的胚胎干细胞至今未成功建系,因此通常采用第二种方式,即对猪体细胞的基因组DNA进行CRISPR/Cas9打靶,并筛选阳性细胞进行体细胞核移植来生产转基因猪[31]。PEF细胞是常用的供体细胞类型,因此,对PEF进行基因编辑是转基因猪生产的第一步。外源基因敲入效率低始终是转基因动物生产的主要问题。研究发现,含有Cas9切割位点的供体质粒可以有效促进外源基因整合至基因组[32]。此外,基于微同源介导的末端连接(microhomology-mediated end joining,MMEJ)的方法,以及基于同源末端连接(homology-mediated end joining,HMEJ)的方法都被证明能显著提高外源基因的敲入效率[33-35]。然而,大多数研究都是在斑马鱼和老鼠身上进行的,而关于猪基因编辑技术的研究较少。
转基因猪在农业和生物医学研究中发挥着重要作用。过氧化物酶体增殖物激活受体γ共激活因子1α(peroxisome proliferator-activated receptor-γ coactivator-1α,PGC1α)转基因猪表现出较高的肉质品质[36]。此外,转黏液病毒抗性(myxovirus-resistant,Mx)基因的猪也被成功用于保护猪免受猪瘟病毒(classical swine fever virus,CFSV)的感染[37]。人白蛋白转基因猪在人类抗病性中发挥重要作用[38]。基因敲入位点的选择对转基因动物的发育和健康具有重要意义,ROSA26是外源性基因敲入的首选位点,其在多个物种中具有保守性[39]。许多外源基因敲入动物通过插入ROSA26位点而成功建立[40]。目前已经确定了猪ROSA26的启动子序列,研究人员发现猪ROSA26的启动子可以通过避免DNA甲基化的方式来驱动基因高而稳定的表达[41]。
尽管转基因猪已经成功生产,但其效率仍然很低。高效的PEF打靶效率是外源基因成功导入的必要条件。目前猪的基因打靶效率在不同的研究中差异较大,Li等[42]针对猪ROSA26第一外显子与第二外显子之间的区域设计了14条sgRNAs,结果发现不同的sgRNA的打靶效率差异较大,且最高的sgRNA打靶效率能达到54.3%,对猪中心体蛋白112(centrosomal protein 112,CEP112)基因的打靶结果也显示最高的打靶效率可达到36.7%[43]。Xie等[39]研究发现,将cas9 mRNA和sgRNA微注射到猪的孤雌胚胎中,经桑格尔测序鉴定的突变效率仅为14.8%,而利用CRISPR/Cas9质粒对PEF细胞进行黑素皮质素受体3(melanocortin receptor 3,MC3R)基因的敲除,也只能实现15.28%的双等位基因突变[44]。以上研究表明,虽然在某些基因的某些位点CRISPR/Cas9的打靶效率已经较高,但对于较难插入外源区域的位点或基因,提高其打靶效率仍然是必要的。目前关于猪ROSA26位点的打靶区域,均设计在第一外显子与第二外显子区域,在本研究中,我们针对猪ROSA26的启动子区进行打靶,并设计了2条靶向猪ROSA26启动子的sgRNAs;利用RGS-CR质粒筛选打靶效率较高的sgRNA。组织特异性启动子可驱动基因在特定组织中表达,而产生组织特异性转基因动物对于基因治疗至关重要[45]。在本研究中,猪ROSA26位点启动子的切割破坏了ROSA26启动子的活性,从而允许外源的组织特异性启动子的敲入,这为后续研究猪的外源基因组织特异性表达提供了参考方向。更重要的是,本研究结果表明,与传统的CRISPR/Cas9质粒相比,利用Cas9/sgRNA体外形成的RNP复合体,可以显著提高sgRNA的打靶效率,为提高后续基因编辑效率奠定基础。
综上所述,本试验成功构建了PX459-cas9-sgRNA和RGS-sgRNA,且sgRNA2的打靶效率高于sgRNA1,且sgRNA2 不存在脱靶效应。1 500 V、3 t、10 ms的电转条件对PEF细胞而言具有较好的转染效果。Cas9/sgRNA体外直接形成RNP的打靶效率比CRISPR/Cas9质粒提高了约1倍,这对于促进基因编辑在猪的应用和提高未来SCNT的效率和外源基因敲入具有重要意义。
猜你喜欢
舰船科学技术(2022年11期)2022-07-15 07:51:56
今日农业(2021年11期)2021-08-13 08:53:24
西藏农业科技(2019年3期)2019-11-04 00:35:10
食品科学(2018年10期)2018-05-23 01:27:28
现代园艺(2018年3期)2018-02-10 05:18:12
上海农业学报(2017年3期)2017-04-10 12:39:12
西南医科大学学报(2015年1期)2015-08-22 13:01:46
中国当代医药(2015年9期)2015-03-01 02:01:59
西南军医(2015年6期)2015-01-23 01:25:50
遗传(2014年3期)2014-02-28 20:58:49
