
苎麻炭疽病病原菌的分离与鉴定
2023-05-24 01:50王绪霞汤涤洛彭洪翠彭蝶黎州刘立军
中国麻业科学 2023年2期
王绪霞,汤涤洛,彭洪翠,彭蝶,黎州,刘立军
(1.武昌首义学院城市建设学院,湖北 武汉 430064;2.咸宁市农业科学院,湖北 咸宁 437100;3.华中农业大学植物科学技术学院,湖北 武汉 430070)
苎麻(Boehmeria niveaL. Gaud)是荨麻科(Urticaceae)苎麻属(Boehmeria)多年生宿根性纤维作物,在我国有着悠久的种植历史。 我国苎麻种质资源和遗传多样性丰富,种植面积和原料产量在世界上处于优势地位[1]。 苎麻炭疽病是苎麻上经常发生的主要病害之一,给苎麻生产带来严重的经济损失[2]。 该病害在我国长江流域以南各省麻区均有发生,发病率达20%~40%,感病后叶片上产生典型病斑进而影响作物生长,发病严重的麻园纤维产量减产达l0%以上[3],并且刮制后的原麻有红褐色斑点,极大影响苎麻纤维的品质[4]。 长期以来,我国苎麻在种质资源创新与鉴定、品种筛选、育种与繁育技术研究[5-8]以及基于UAV-RGB 遥感系统估算苎麻产量等方面均取得重大进展[9-10],但在苎麻炭疽病病害及其病原菌的研究方面还比较薄弱。 1914 年日本学者Sawada 在中国台湾首次对该病菌进行分离和形态学观察,并将苎麻炭疽病菌命名为C. boehmeriaeSawada(以寄主来源和其名字命名)[11-12]。 二十世纪八九十年代,我国苎麻研究专家对苎麻炭疽病的症状及各个生育期病害发生的特点、侵染源、发生规律等进行了系统性研究,提出了苎麻炭疽病的防治措施,为苎麻炭疽病害的防治提供了指导[3-4,13-14]。 至今,国内外关于苎麻炭疽病病原菌的研究报道仍较少。 在以往苎麻炭疽病的田间调查、病害防治、苎麻品种抗性检测和对苎麻炭疽菌的研究报道中,根据寄主来源和发现者命名的“Colletotrichum boehmeriaeSawada”一直作为苎麻炭疽病菌的病原,但其不能说明苎麻炭疽菌真正的病原和分离地位。 准确鉴定引起苎麻炭疽病的病原菌是研究病菌生物学特性、发生规律和开展病害防治的基础。
本研究通过采集苎麻炭疽病感病的叶片,采用传统组织分离法进行病原真菌的培养和分离,依据病菌的形态学特征、致病性测定、分子生物学和形态学结合的方法对引起苎麻炭疽病的病原菌进行鉴定,旨在明确引起苎麻炭疽病的病原菌。
1 材料与方法
1.1 试验材料
从湖北省武汉市华中农业大学苎麻材料圃、湖北省咸宁市苎麻基地大田、中国农业科学院麻类研究所苎麻材料圃(湖南长沙)和江西省宜春市农科所苎麻材料圃等地,采集症状典型的苎麻炭疽病病叶,用于病原菌的分离。
1.2 试验方法
1.2.1 病菌分离与纯化
应用常规植物病原菌的分离法进行病菌的分离[15-16]。 在无菌操作台上,切取病健交界处约2 mm×2 mm 的病组织,先用灭菌水冲洗干净,再用75%酒精消毒1 min,5%次氯酸钠消毒3 min,最后用无菌水冲洗3 遍,将消毒好的病组织置于PDA 平板上,28 ℃下暗培养3~4 d。 待菌落长出后,重复纯化2~3 次。 将分离的菌株保存在PDA 平板中,4 ℃储存备用。
1.2.2 致病性检测
分生孢子液接种:选取新鲜健康的苎麻叶片(华苎4 号),放入铺有吸水纸的盘中摆放整齐,吸水纸经灭菌水滋润。 在每个叶片上滴加分生孢子液,每片叶片接10 处,每处接种6 μL(1×106个/mL),3 个重复,以清水作为对照组,用保鲜膜封盘并置于28 ℃恒温培养箱中12 h 光暗交替培养,连续观察10 d,记录叶片发病情况。
菌丝块接种:叶片选取同以上分生孢子液接种。 PDA 平板上活化好的菌落用打孔器(d=6 mm)打取菌丝块。 将菌丝块面朝下接种到叶片上,每片叶片接5 ~6 个菌丝块,3 个重复,清水作为对照,培养条件和观察方法同分生孢子液接种。 叶片发病后从病斑上再次分离病原菌,比较再分离菌株与接种菌株的一致性,完成柯赫氏法则的验证。
1.2.3 病原菌的鉴定
形态学鉴定:将菌丝块接种到PDA 平板上,在28 ℃培养箱里黑暗培养,3 d 后观察并记录菌落特征,主要包括菌落颜色、形态和生长情况。 在显微镜下观察分生孢子、附着胞及孢子梗的形态,用显微镜及标尺测量分生孢子的大小,并计算分生孢子的长宽比例,查阅真菌分离鉴定工具书,初步判定病菌的种类。
分子生物学鉴定:采用CTAB 法提取病原菌DNA,用真菌核糖体内部转录序列ITS 通用引物ITS1(5’-TCCGTAGGTGAACCTGCGG-3’)和ITS4(5’-TCCTCCGCTTATTGATATGC-3’)[17]、β-微管蛋白基因(β-tubulin,TUB2)的PCR 扩增引物T1(5’-AACATGCGTGAGATTGTAAGT-3’)和βt2b (5’-ACCCTCAGTGTAGTGACCCTTGGC-3’)[18]对目的基因区域进行PCR 扩增。 将PCR 产物纯化后进行T 载体的连接和大肠杆菌的转化,并对质粒进行测序。 将所测得的序列在NCBI 网站(http:/ /www.ncbi.nlm.nih.gov)进行同源性比较,以近缘菌株构建系统发育树,确定其分类地位。
2 结果和分析
2.1 苎麻炭疽病的田间症状
根据苎麻炭疽病田间症状观察,从我国3 个省份(湖北、湖南、江西)主要苎麻产区采集具有苎麻炭疽病症状的叶片。 对田间症状和采集的叶片进行观察,发现苎麻炭疽病主要症状包括:染病叶片呈现典型的圆形病斑,病斑直径1~3 mm,病斑边缘黑色,中央褐色,可扩展,严重的呈中间凹陷坏死症状(图1-A~C);叶片上具有大量小圆斑症状,病斑直径<1 mm,病斑中央灰白色,边沿深褐色,扩展有限(图1-D);叶片上具有大量小圆斑症状,病斑直径<1 mm,可扩展成片(图1-E);叶片上具有黑色大斑,直径>5 mm,病斑扩展成片(图1-F)。

图1 苎麻炭疽病田间叶片症状Fig.1 Representative symptoms of ramie anthracnose on leaves in filed
2.2 苎麻炭疽病病原菌致病性测定结果
本试验共分离89 个菌株,选取形态有差异且能在PDA 培养基上直接产生或者切断菌丝后能诱导产生橘红色分生孢子的23 个菌株进行致病性测定。 先采用分生孢子接种法对菌株的致病性进行测定,结果显示,菌株大致有强致病性、弱致病性和无致病性3 类(图2)。 23 个菌株中有14个菌株可以引起“华苎4 号”发病,其中,强致病性菌株占总测试菌株的61%。 强致病性菌株接种叶片3 d 后,叶片上接种点开始出现病斑,随着培养时间的延长病斑变大,扩大成片,最后整个叶片退绿,腐烂(图2-A1~A4);第二类弱致病性菌株,接种叶片3 d 后出现病斑,随着培养时间的延长,坏死部位没有继续恶化,10 d 后叶片仍然完好(图2-B1 ~B4);第三类菌株接种后叶片没有受感染,叶片表面未见任何病斑(图2-C1~C4 )。

图2 苎麻炭疽病菌分生孢子测定致病性Fig.2 Pathogenicity test of pathogens of ramie anthracnose with conidia
将致病性强的菌株用菌丝片法再次进行致病性检测。 结果显示,接种3 d 后接种处开始发病,初为褐色斑点(图3-A),接种7 d 后病斑颜色加深并扩展为圆形,有的病斑连成片导致叶片腐烂(图3-B)。 从病组织再分离的病原菌与接种用的菌株形态上一致。
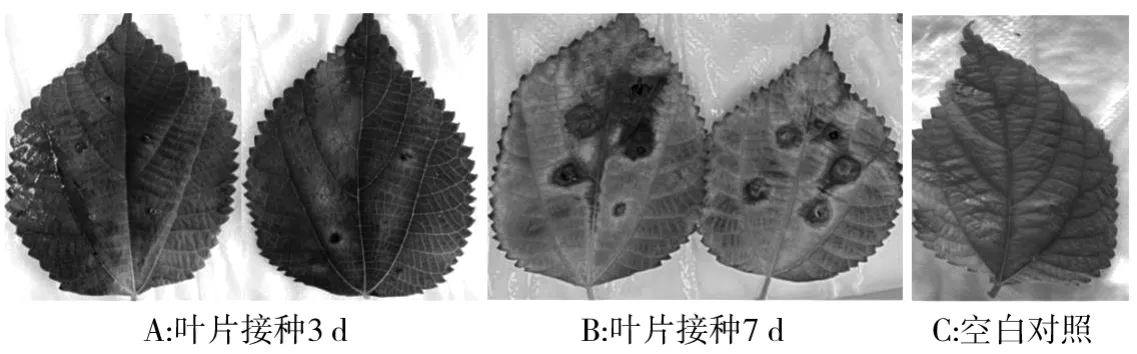
图3 苎麻炭疽病菌菌丝块测定致病性Fig.3 Pathogenicity test of pathogens of ramie anthracnose with mycelium
2.3 苎麻炭疽病菌的形态特征及分类地位
2.3.1 形态学鉴定结果
苎麻炭疽病菌在PDA 培养基上菌落呈圆形,菌落边缘整齐。 菌丝匍匐状,稀疏至茂密,大部分菌株菌丝最初为白色,随着培养时间的延长慢慢变为淡灰色,有的是褐色、墨绿色(图4-A1、B1、C1、D1)。 分生孢子橘红色,有的点状分布,有的成片分布,分生孢子单胞,无色透明,卵圆形,有的内含油球(图4-A2、B2、C2、C3、D2)。 有的菌株具黑色或者褐色刚毛,有分隔,顶端尖锐(图4-D3)。 分生孢子梗簇生或单生,无色或淡褐色,直或弯曲,具分隔,基部略膨大。 菌丝有隔,直径2.55~3.83 μm,平均3.01 μm。 孢身大小及孢子长宽比例见表1。 查阅真菌鉴定手册,根据病菌形态特征,初步判定苎麻炭疽菌病原为胶孢炭疽菌C. gloeosporioides(胶孢炭疽菌刚毛少,分生孢子圆筒形,(11~18)μm×(4~6)μm)和希金斯炭疽菌C. higginsianum(希金斯炭疽菌分生孢子盘上有刚毛数根,分生孢子长椭圆或圆筒形,两端钝圆,无色,单胞,(15~21)μm×(3.0~5.5)μm)。
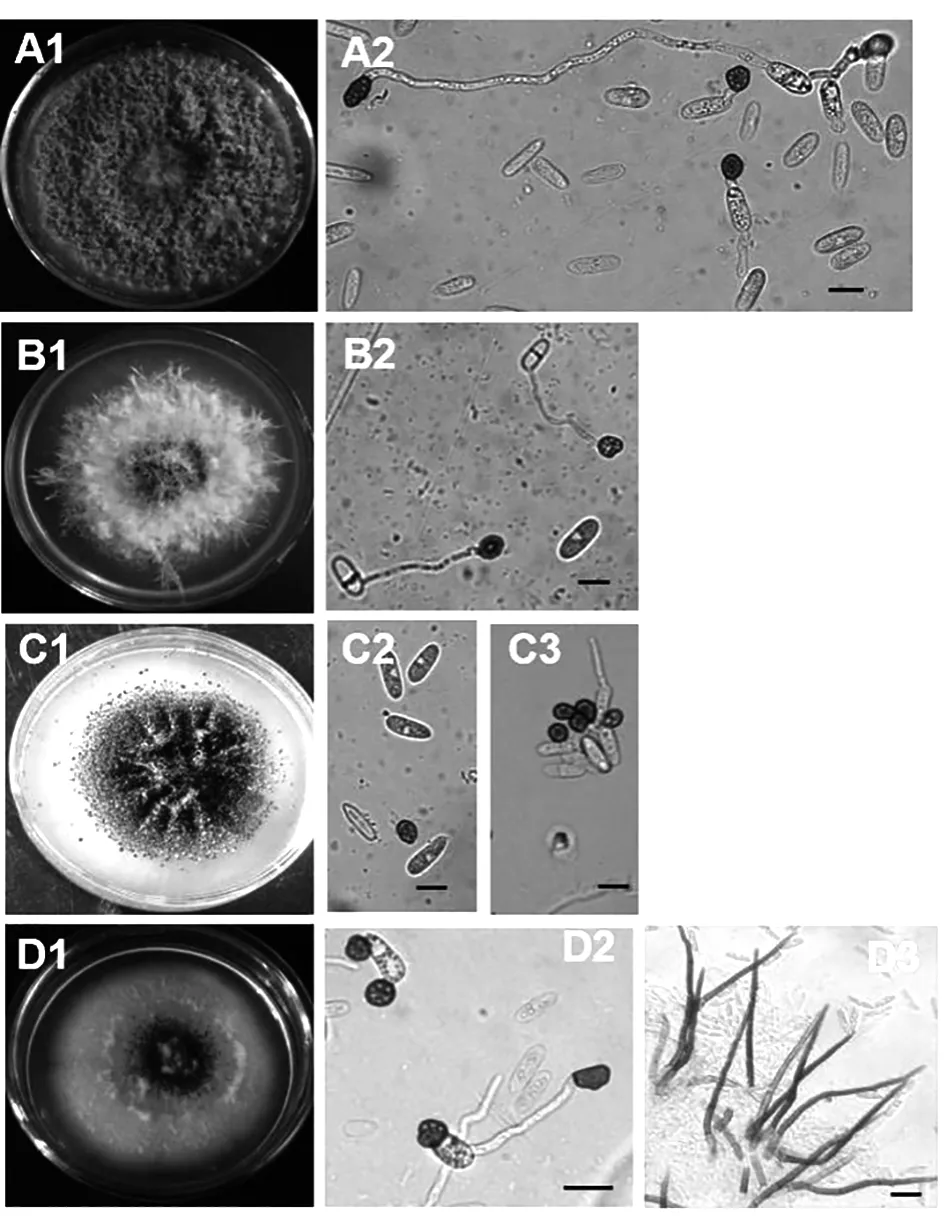
图4 苎麻炭疽病病原菌形态特征Fig.4 Morphylogy of pathogens of ramie anthracnose
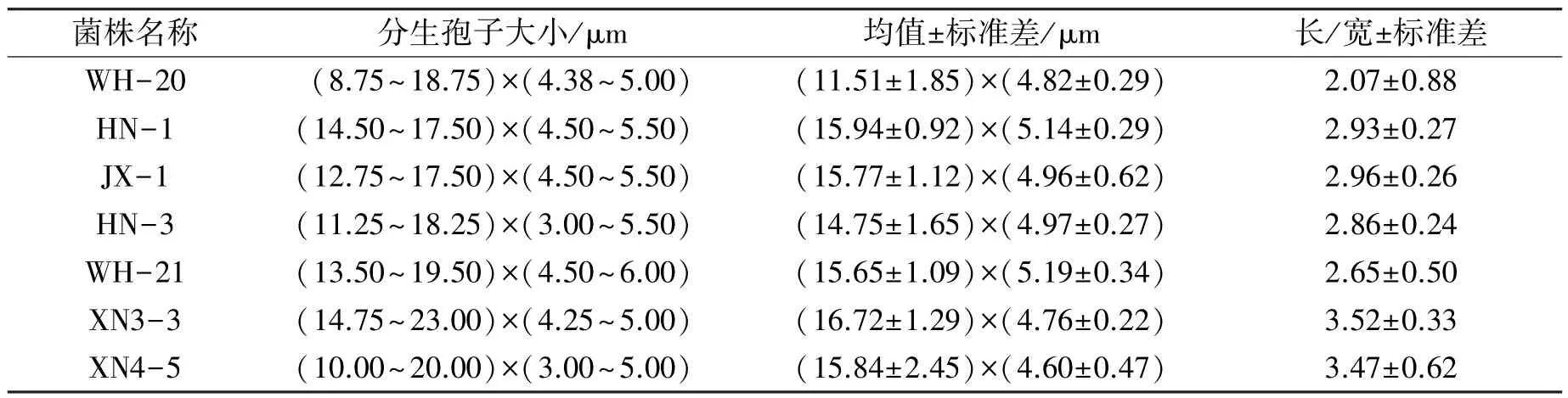
表1 苎麻炭疽菌代表性菌株分生孢子大小测定Table 1 Conidia sizes of pathogens of ramie anthracnose
2.3.2 分子生物学鉴定结果
选取具有正常致病力且经过柯赫氏法则验证的14 个菌株进行ITS基因克隆和测序,将分离菌株的测序结果在GenBank 中进行BLAST 比对。 比对结果显示,菌株与胶孢炭疽菌C. gloeosporioides、果生炭疽菌C. fructicol、暹罗炭疽菌C. siamense和希金斯炭疽菌C. higginsianum的同源性达97%~99%(Accession number:FJ515005、FJ459930、HM016798、AB042317.1、GU935872.1)。 而且序列比对中发现,C. gloeosporioides、C. fructicola、C. siamense和C. higginsianum这4 类植物炭疽菌病原的ITS序列本身同源性高达99%,单通过ITS基因无法将菌株鉴定到种。
进一步对测试菌株进行β-tubulin基因的克隆。 表2 中列举了菌株的来源和β-tubulin基因在NCBI 上的登录号。 将分离菌株的β-tubulin基因测序结果在GenBank 中进行BLAST 比对,查找相似物种的β-tubulin序列,使用MEGA 7.0 最大似然法对相似性较高的菌株及同属不同种菌株进行分子系统发育分析(图5)。 结果显示,从湖南采集的菌株中鉴定出3 个炭疽菌属的病菌:C. fructicola、C. siamense和C. gloeosporioides;湖北武汉的材料中包括种C. fructicola和C. gloeosporioides;江西的菌株中,并未分离到C. gloeosporioides,分离的菌株为C. fructicola和C. siamense。 采自湖北咸宁的材料,通过β-tubulin比对,与人参生刺盘孢C. panacicola和希金斯炭疽菌C. higginsianum的同源性都为99%,而这两个病原菌属于C. destructivumspecies complex 复合群,所以分子序列上同源性很高。 真菌鉴定手册记载希金斯炭疽菌C. higginsianum分生孢子盘上有刚毛数根,分生孢子圆筒形,(15~21)μm×(3.0~5.5)μm。 本试验中分离自咸宁的菌株其分子孢子特性(图4-D1~D3,表1)与C. higginsianum的描述一致,再结合分子检测的结果,判定分离自咸宁的菌株属于C. higginsianum。
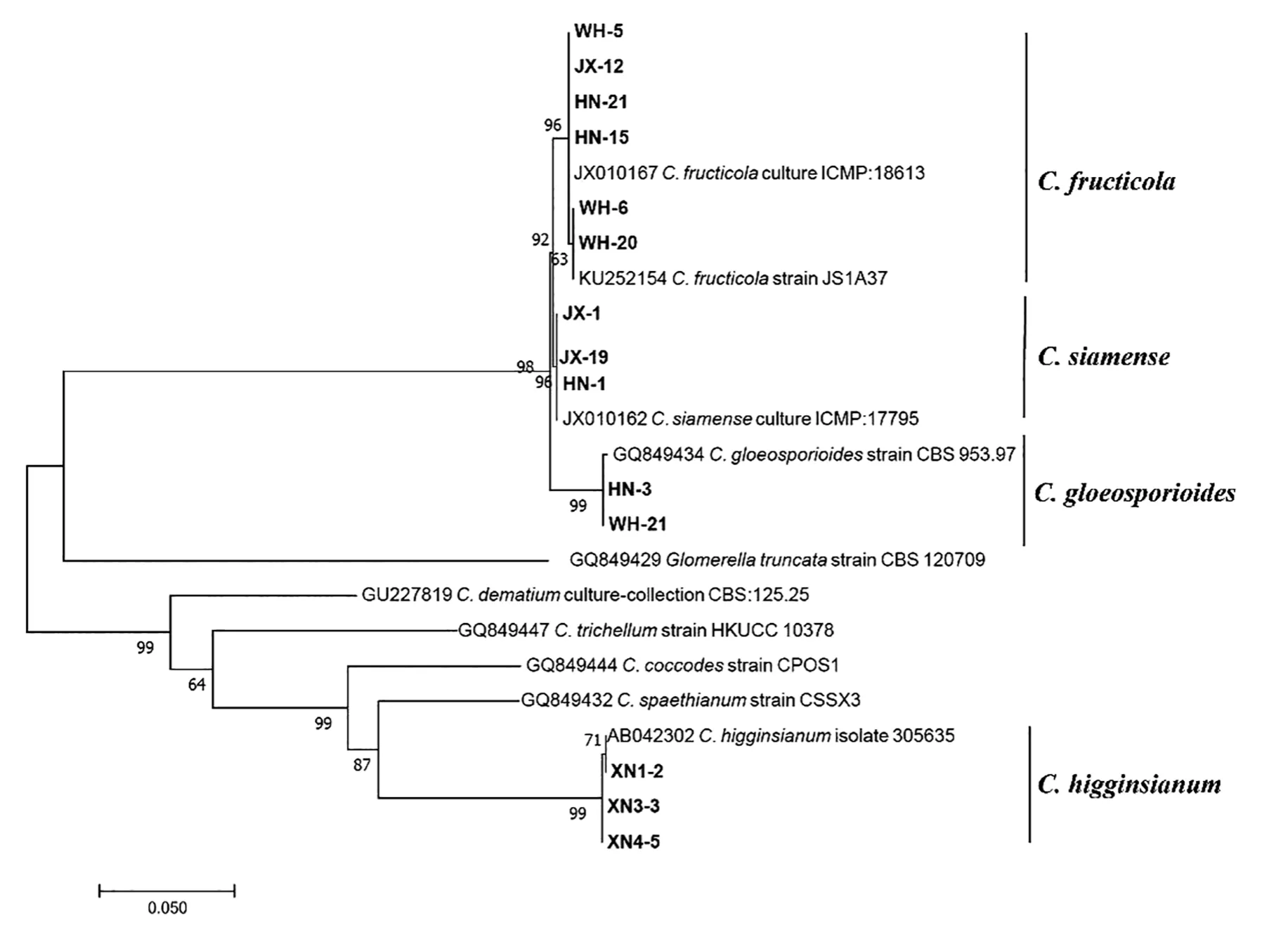
图5 基于β-tublin 基因序列构建的苎麻炭疽菌菌株系统发育进化树Fig.5 Phylogenetic tree of ramie anthracnose strain based on β-tublin genes sequences

表2 基于β-tubulin 基因的系统发育分析所用菌株信息Table 2 Informations used for phylogenetic analysis based on β-tubulin genes
3 讨论与结论
苎麻炭疽病由植物炭疽菌属(Colletothrichcumspp.)真菌引起。 该属真菌是一类全球性分布的重要植物病原菌,被列为全球第八大植物病原真菌[19]。 目前已报道约176 属190 余种植物可被炭疽菌侵染[20]。 炭疽菌类真菌命名方式和系统分类方法发生过数次重大的变更,存在种间性状交叉和系统发育关系不清的问题。 1831—1957 年,其分类依据主要以寄主范围为主,即只要从一种尚未报道的寄主上分离获得该属真菌,即被视为新种。 该时期至少750 个新种被描述,导致许多物种同物异名。 Von Arx 发现该属真菌分类混乱,依据分生孢子和产孢细胞等显微形态特征,并结合纯培养特征,将750 多个种合并为11 个种[21]。 Sutton 在Von Arx 的分类基础上,于1962—1992 年,以分生孢子和附着胞形态特征,结合纯培养物特征和寄主范围,把该属扩展到包含部分复合种的39 个种[22]。 2009—2019 年,Damm 等[23-24]分别对存在争议的种的混乱问题又进行整理。 目前,炭疽菌属共包括14 个复合种,其中C. gloeosporioides复合种包括38 个近似的物种[25],近年又扩展至22 个复合种[26]。
准确区分和鉴定病害的病原菌是制定病害有效防御策略的基础。 对病原真菌的鉴定,常以纯培养物上产生的分生孢子、附着胞、分生孢子梗和产孢细胞的形态和大小等特征进行形态学鉴定[27]。 对于特征明显的炭疽菌种采用形态学能够容易被鉴定出来,但是某些近似炭疽菌种却难以准确区分,比如Liu 等[28]认为胶孢炭疽菌C. gloeosporioides、暹罗炭疽C. simense和果生炭疽菌C. fruticola等一些近种,其菌落形态存在细微差异,单依据分生孢子形态和大小很难明显区分,并且一些菌株形态学特征不够稳定,易受培养条件的影响。 真菌的分子鉴定是从基因水平上反映菌株间遗传特性,DNA 序列特征不会随环境的改变而变化,特异性高,准确可靠,借助分子手段进行辅助鉴定可以弥补形态学鉴定的不足[29]。 除病毒以外,所有生物都具有核糖体基因转录间隔区(ITS序列)区段的基因,该序列种间高度保守,可作为研究生物系统进化及分类的依据[30]。 另外基于胶孢炭疽菌C. gloeosporioides复合种的病原种类多,单选用ITS序列可能无法对小种进行有效的区分。 目前,基于多基因序列分析,将ITS基因、肌动蛋白基因(Actin gene,ACT)[30]和β-微管蛋白基因(β-tubulin,TUB2)[31]、几丁质合成酶A 基因(chitin synthase A gene,CHS1)等[32]序列进行联合分析用于炭疽菌的分子鉴定,能较好地区分炭疽菌属真菌的大部分种。
本研究对采集自我国苎麻主产区的苎麻炭疽病病株进行病原菌的分离,首先通过形态学对分离的病菌进行分析,初步判定引起苎麻炭疽病病原是刺盘孢菌属中的胶孢炭疽菌C. gloeosporioides。 通过对ITS序列的克隆发现胶孢炭疽菌C. gloeosporioides、果生炭疽菌C. fructicol、暹罗炭疽菌C. siamense和希金斯炭疽菌C. higginsianum4 类炭疽菌的ITS序列本身有很高的同源,甚至同源性达99%,单通过ITS基因无法将菌株进行鉴定到种。 所以通过本研究可以推断ITS序列无法对引起苎麻炭疽病的病原菌鉴定到种。 进一步通过微管蛋白β-tubulin基因的克隆和肌动蛋白基因(Actin gene,ACT)(结果未显示)的克隆多基因联合分析,结果确定引起苎麻炭疽病的病原菌为胶孢炭疽菌C. gloeosporioides[33]、果生炭疽菌C. fructicola、暹罗炭疽菌C. siamense和希金斯炭疽菌C. higginsianum[34]。 微管蛋白β-tubulin基因的克隆和肌动蛋白基因(Actin gene,ACT)可以对苎麻炭疽菌进行小种间的鉴定。 依据新的炭疽菌种的分类方法,胶孢炭疽菌C. gloeosporioides、果生炭疽菌C. fructicola、暹罗炭疽菌C. siamense隶属于胶孢炭疽菌复合群C. gloeosporioidescomplex,希金斯炭疽菌C. higginsianum属于毁灭炭疽菌C. destructivumcomplex 复合群。
本研究对我国苎麻主产区湖北省、湖南省和江西省的苎麻炭疽病病原菌进行了分离和鉴定,为病菌生物学特性及其致病基因的研究奠定了基础。 鉴定出的4 类病原菌在苎麻上引起病害的发生规律以及各苎麻生长地区中优势种群和病菌的致病基因、致病力分化、遗传多样性等还需要进一步研究和分析。 对病原菌进行基因组测序,利用生物信息学方法进行组学序列分析、基因功能注释和比较基因组学分析,是研究植物病原菌间基因变异重组、突变以及插入/缺失、遗传多样性、致病力分化机制的有效方法[35]。 全基因组分析已用于柑橘溃疡病菌、水稻稻瘟病菌、芽孢杆菌、芒果炭疽菌等多种病原菌的基因组分析。 大量病原菌全基因组测序及比较基因组研究文献和炭疽菌全基因组序列数据,将有助于苎麻炭疽菌基因组测序、组装和功能基因的预测。
猜你喜欢
今日农业(2022年15期)2022-09-20
中国森林病虫(2021年6期)2021-12-20
现代畜牧科技(2021年4期)2021-07-21
中国森林病虫(2018年4期)2018-09-19
动物营养学报(2017年2期)2017-02-28
中国麻业科学(2015年5期)2015-12-28
中国麻业科学(2015年5期)2015-12-28
塔里木大学学报(2015年1期)2015-04-25
营销界(2015年23期)2015-02-28
中国麻业科学(2014年5期)2014-12-05
