
Mitochondrial dysfunction in glaucomatous degeneration
2023-05-15 09:20:38ZiQiaoZhangZhiXieSenYuanChenXuZhang
Zi-Qiao Zhang, Zhi Xie, Sen-Yuan Chen, Xu Zhang
1Affiliated Eye Hospital of Nanchang University, Nanchang University School of Ophthalmology & Optometry, Jiangxi Provincial Key Laboratory for Ophthalmology, Jiangxi Research Ⅰnstitute of Ophthalmology & Ⅴisual Science,Nanchang 330006, Jiangxi Province, China
2Queen Mary School, Nanchang University, Nanchang 330006,Jiangxi Province, China
Abstract
INTRODUCTION
G laucoma is a group of neurodegenerative diseases[1]marked by the irreversible death of retina ganglion cells (RGCs), characteristic optic atrophy, and loss of visual fields[2].At present, glaucoma has become the leading cause of permanent blindness all around the world[3].A comprehensive assessment from 2014 anticipated that there will be 111.8 million glaucoma patients worldwide in 2040[4].Based on whether the anterior chamber angle is open or not and whether the intraocular pressure (IOP) is raised, glaucoma can be roughly divided into three categories: open angle glaucoma(OAG), angle-closure glaucoma (ACG) and normal tension glaucoma (NTG)[5].In light of the similarities between glaucoma and primary mitochondrial optic neuropathies[6-7],numerous studies have concentrated on the impact of mitochondrial dysfunction in the course of glaucoma, which is the main topic of our review as well.
Although glaucoma has been a threat to human health for many years, the exact pathogenesis and contributing factors of glaucoma remain an open question to be elucidated.On the one hand, it is acknowledged that the primary risk factor for glaucoma is pathologic elevated IOP[8].The stability of IOP is kept by the aqueous humor generated by the ciliary body.When aqueous humor is generated too much or outflow is blocked, the IOP will rise progressively.Increased IOP causes mechanical stress and injury to RGCs and the optic nerve head (ONH), particularly the lamina cribrosa and surrounding tissues[9].Damaged optic neuronal axons under prolonged stress result in poor transport, impeding the retrograde movement of essential trophic factors from the brainstem to the RGCs[10].Meanwhile, a decrease in the transfer of oxygen and nutrients to the retina and optic nerve is generated as a result of blood vessel compression brought on by high IOP,which causes the production of reactive oxygen species (ROS)and oxidative damage[11].
Aging is another key factor affecting the progression of glaucoma[8].Although there are significant differences in glaucoma prevalence of different races, it increases substantially with age[12].What’s more, our group has shown that mitochondrial DNA (mtDNA) integrity, the copy number of mtDNA, fusion mediators, mitophagy, and genes related to antioxidation all show a downward trend with the increase of age in the retina of zebrafish models[13].Additionally, due to the increasing need for light-emitting diodes (LEDs) and digital screens, light damage is a significant factor in ocular aging.Exposure to certain wavelengths or intensities of light for a certain time can result in serious damage to the retina[14-15], which may facilitate the development of age-related ocular disorders.Other risk factors[16], including thin corneas, positive family history,and ethnicity, are also connected with the pathogenesis of glaucoma.
Currently, IOP is the sole variable that can be controlled and is proven to be effective in glaucoma treatment[17].However, the degeneration of RGCs and axons is still progressing after strict control of IOP[7].Therefore, finding the exact pathological mechanisms of glaucoma and developing specific treatment options are urgently needed.Here we review the significance of preserving healthy mitochondrial function for the retina and optic nerve and how mitochondrial dysfunction affects the pathogenesis of glaucoma.Then we summarize current and potential therapeutic options, particularly mitochondrial-related ones.The therapies aimed at improving mitochondrial function at the cellular or molecular level may offer new hope for research and clinical utility to delay the process of glaucoma.
Normal Mitochondrial Functions in the Retina and Optic Nerve
Energy and reactive oxygen speciesIn normal situations,the light passing through the pupil is sensed by retinal photoreceptor cells[18].The signal will be transmitted by a series of retinal cells and finally by RGCs to the visual cortex.The axons of RGCs forming the optic nerve cross the lamina cribrosa and are wrapped in myelin sheaths post the lamina cribrosa[19].Instead of carrying out saltatory conduction,unmyelinated prelaminar and laminar RGC axons transmit electronic signals by high-density voltage-sodium channels and Na+-K+adenosine triphosphate (ATP) pumps[19].Because of this special structure, the neurons and optic nerve require oxidative phosphorylation (OXPHOS) to produce large quantities of ATP to keep the electrochemical gradients for transmitting signals[20].Corresponding to this condition, a physiological rise in the number of mitochondria is found in the unmyelinated RGC axons[21].Mitochondria are important sites for producing energy in the form of ATP[22].Meanwhile, it participates in cell apoptosis, the balance between the production and removal of active substances, and the regulation of intracellular calcium absorption and release[23-24].As the primary energy source of most cells, the mitochondrial OXPHOS system includes two essential components – the electron transport chain (ETC) and ATP synthases (Figure 1).Five protein complexes helping complete the process of OXPHOS are located on the inner membrane of mitochondria[25].Electrons from niacinamide adenine dinucleotide (NADH) or reduced flavin adenine dinucleotide (FADH2) are sequentially transferred to oxygen through the complexes, while protons are pumped into the space between the inner and outer membranes to create a proton concentration gradient[26].When these protons return to the mitochondrial matrix, ATP is produced by ATP synthase[26].The stable production of ATP in mitochondria is the basis for the normal operation of various cellular functions.
It is worth mentioning that the mitochondrial respiratory chain provides sufficient energy but also produces reactive oxygen or nitrogen species (ROS/RNS; Figure 1), such as hydrogen peroxide (H2O2), hydroxyl radical (OH), the anion radical (O2-)and nitric oxide radical[27-28].Complex I and III of the ETC are the most active sites for the generation of ROS[29].Despite the fact that ROS and RNS, which is generated by the reaction of nitric oxide and ROS, can act as critical second messengers in many signaling processes[7,30], excessive reactive species can oxidize DNA and various biological molecules, representing the primary cause of damage to cellular structures[30].There are a lot of antioxidants that fight against oxidation inside cells,such as glutathione peroxidases, superoxide dismutase, and catalases[31], which can alleviate the harmful effects resulting from reactive species.Nevertheless, under some situations and damage, the production of reactive substances surpasses the capacity of the cellular antioxidant mechanism[28].This leads to the state of oxidative stress, which is regarded as an imbalance between oxidants and antioxidants that tends to favor oxidation, resulting in destroy of redox signaling and/or molecular damage[27,32].Oxidative stress can irreparably damage mitochondrial and cellular functions.

Figure 1 Electron transport chains in healthy and dysfunctional mitochondria The ETC is made up of four different protein complexes:complex I (NADH dehydrogenase), complex II (succinate dehydrogenase), complex III (cytochrome reductase), and complex IV (cytochrome oxidase).Electrons that originate from NADH or FADH2 are finally transported to oxygen after passing through complex I or complex II,coenzyme Q, complex III, cytochrome c, and complex IV.During the process, oxygen can react with electrons in advance to produce ROS.In healthy mitochondria, ROS can be well removed while in dysfunctional mitochondria, the antioxidant capacity of mitochondria and the electron transport efficiency are reduced, resulting in excessive ROS and causing oxidative damage to mitochondria[32].ETC: Electron transport chain;ROS: Reactive oxygen species; Cyto c: Cytochrome c; Q: Coenzyme Q.Created by Figdraw.
Mitochondrial Quality ControlAs byproducts of mitochondrial energy production, ROS can cause mtDNA mutations and interfere with protein folding and structure.Therefore, a sophisticated quality control mechanism needs to be developed to remove damaged components and biosynthesize new macromolecules[33].In this regard, it is essential to mention mitophagy as well as mitochondrial dynamics ‐ fusion and fission.Mitophagy, called the selective autophagy of mitochondria[34], is a mechanism that eliminated damaged mitochondria and is precisely mediated by PTENinduced putative kinase 1 (Pink1) and Parkin[35].In addition,the morphology and connectivity of the mitochondrial network are controlled by the processes of mitochondrial fusion and fission, depending on the demand for specific biological needs[36-37].Fusing together several mitochondria to produce a single mitochondrion is an example of fusion while fission refers to the process of breaking apart a single large mitochondrion into many smaller ones[38].Mitochondria constantly undergo the cycle of fusion and fission, so they come in a variety of shapes[39].Fusion mediated by optic atrophy 1 (Opa1) and mitochondrial fusion proteins 1/2 (Mfn1/Mfn2) can dilute mutant mtDNA with non-mutant mtDNA for mitigating DNA damage[40].In an obvious fusion event, two mitochondria collide end to end and fuse at the site of the collision[39].The outer membrane of mitochondria fuses first and the inner mitochondrial membrane (IMM)fuses as well.Meanwhile, the mitochondrial contents mix and spread throughout the new mitochondria.In addition,there is a course called the “kiss-and-run fusion”event[41].One mitochondrion does not undergo an obvious structural combination, but exchanges matrix contents with another during brief encounters[41].Therefore, the significance of mitochondrial fusion is not only morphological regulation but also content exchange.As for the fission event, it contributes to biosynthesizing new mitochondria and also to removing dysfunctional mitochondria[42-43].How this dual fate happens has intrigued scientists.New studies find that when cells are about to proliferate, mitochondria tend to undergo midzone fission[44].However, before most peripheral fission, which is mediated by dynamin-related protein 1 (Drp1) and fission protein 1[45], mitochondrial membrane potential and proton motive force are reduced, while ROS and calcium levels are increased.The cellular changes suggest that mitochondrial dysfunction may drive mitochondria to undergo peripheral fission to remove damaged mitochondria[44].Although it is not clear how to keep the balance between fusion and fission,it has been found that fission usually occurs shortly after fusion[42](Figure 2).After a fission event, some mitochondria remain in a state of constant depolarization suggesting dysfunction.Such mitochondria are solitary for several hours in the pre-autophagic pool and then cleared by mitophagy[42].After undergoing a brief period of depolarization, healthy mitochondria return to normal membrane potential and continue the cycle of fusion and fission[42].
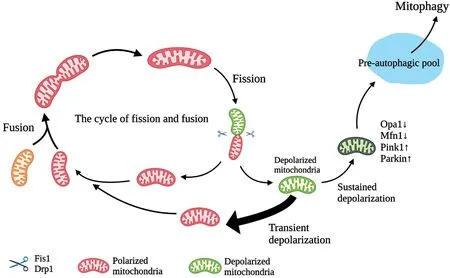
Figure 2 A schematic illustration of the relationship between mitochondrial fission, fusion, and mitophagy Mitochondria are continuously recycled between mitochondrial fission and fusion events.Mitochondrial fusion mechanisms dilute the effects of mutation by fusing mutated mtDNA (orange) with non-mutated mtDNA.When the damage exceeds a certain limit, mitochondrial fission occurs, in which mitochondria depolarize.Subsequently, these mitochondria either return to normal membrane potential (thick arrow) and continue the cycle, or remain depolarized[42].However, sustained depolarization will trigger Opa1 cleavage and Mfn1 reduction, along with Pink1/Parkin accumulation.Eventually, these mitochondria are cleared by mitophagy after remaining in the pre-autophagic pool for several hours.mtDNA: Mitochondrial DNA; Opa1: Optic atrophy 1; Mfn1:Mitofusin-1; Pink 1: PTEN-induced kinase 1.Created by Biorender.
Mitochondrial Dysfunction in GlaucomaMitochondrial dysfunction means that mitochondria are not able to carry out normal functions.Because of the high-energy demand,it is essential that the neurons in the retina and optic nerve have the appropriate number of mitochondria and that these mitochondria are functioning properly[23].The aspects of mitochondrial dysfunction can be described by crosstalk among oxidative stress, membrane depolarization, mtDNA damage, reduction of ATP synthesis, cristae decomposition,and defective mitophagy[11,35], all of which can be reflected in the course of glaucoma.Here we mainly summarize mitochondrial dysfunction into three major blocks for review:mtDNA damage, oxidative stress, and defective mitochondrial quality control.
Mitochondrial DNA damageImportantly, mtDNA is close to the location of the mitochondrial respiratory chain and not safeguarded by protective proteins[32], making it exceptionally susceptible to oxidative damage caused by ROS.Hence, the rate of mutation in mitochondrial DNA is significantly higher than that of nuclear DNA[35].ROS can cause single-strand breaks (SSBs), double-strand breaks (DSBs), or oxidized bases[46], resulting in mtDNA mutations.A previous study has shown that the integrity and volume of mtDNA gradually decline with age on account of the accumulation of oxidative damage from ROS[47].The extent of the damage can be measured by the level of a biomarker called 8-hydroxy-2’-deoxyguanosine (8-OH-dG)[48], which indicates oxidative damage to DNA.The contents of 8-OH-dG in the aqueous humor of primary open angle glaucoma (POAG) patients are considerably greater than that of the healthy group[49].Besides, the elevated level of 8-OH-dG was discovered in the trabecular meshwork (TM) of POAG patients, which suggests that the damage to mtDNA is not confined to the retina but is widespread in the eyes.
What’s more, a previous study showed that 27 different novel mtDNA nonsynonymous mutations are identified in POAG patients, among which 22 mutations could be pathogenic[50].Although mtDNA encodes only about 1% of mitochondrial proteins, it contains protein subunits involved in OXPHOS[35].Mutations of related genes or defects in components of OXPHOS could contribute to mitochondrial dysfunction.For instance, mutations in the mitochondrially encoded cytochrome B (MT-CYB), which encodes cytochrome b in Complex III, can cause defects in Complex III and disrupts the production of ATP[51].In TM cells, the complex I inhibitor Rotenone could increase the generation of ROS and induce the release of cytochrome c, finally leading to oxidative stress and cell apoptosis[52].Thus, damaged mtDNA can cause negative consequences for ROS production and clearance, ATP production, and other mitochondrial functions[47].

Figure 3 A schematic diagram of the relationship between oxidative stress and high IOP Excessive ROS can damage the TM cells, greatly increasing their resistance to aqueous humor and making persistent high IOP.High IOP causes mechanical injury to the optic nerve and compresses the retinal central artery, resulting in reduced blood flow and oxygen deprivation.Hypoxia reduces ATP production and stimulates the generation of superoxide by complex III.It also results in decreased mitophagy, which further intensifies oxidative stress.IOP: Intraocular pressure; ROS: Reactive oxygen species; TM:Trabecular meshwork; ONH: Optic nerve head.
Oxidative stressOxidative stress is an essential component in the course of glaucoma, particularly in the occurrence of mitochondrial dysfunction.According to the present studies,oxidative stress can be a cause and/or a concomitant result of mitochondrial dysfunction.The specific mechanisms need to be further explored and summarized, which is the focus of discussion in this section.
Intraocular hypertension and oxidative stressBased on the previous studies, the abnormal structure and function of TM cells could be a crucial reason for increased IOP in primary OAG[48](Figure 3).Due to the fact that the TM has a lower level of antioxidant defense than both the cornea and iris, TM cells are the most vulnerable tissues to oxidative damage in the anterior chamber[53].The imbalance between oxidation and antioxidation in aqueous humor leads to the increase of ROS,which lowers the adherence of TM cells to the extracellular matrix and stimulates the pathway of nuclear factor kappa-B(NF-κB) in addition to damaging the mtDNA of TM endothelial cells, eventually leading to the collapse, fusion and enlargement of TM cytoskeleton[48].NF-κB is a kind of transcription factor whose constant activation can induce the expression of proinflammatory factors[54], aggravating the inflammation responses in glaucoma.All of these changes deteriorate the abnormal resistance of TM cells to aqueous humor drainage, giving rise to progressively increasing IOP.
The gradual rise in IOP causes mechanical injury to the ONH and surrounding tissues[55], thereby compressing the central retinal artery.Ⅴascular dysregulation interferes with the self‐regulatory function of ocular perfusion, so the eyes can no longer maintain a constant blood flow.This impaired blood flow to the eye results in a decreased supply of oxygen to the retina.Ⅴascular dysfunction allows ocular structures, including the retina and TM, to undergo repeated reperfusion injury and unstable oxygen supply[56], exacerbating oxidative stress and poor availability of energy within cells.During glaucoma,RGC axons primarily rely on glycolysis to compensate for deficient mitochondrial function, but glycolysis is not sufficient to salvage the metabolic vulnerability caused by glaucoma[57].When the retina is hypoxic-ischemic, three types of nitric oxide synthases are produced: neuronal (nNOS), endothelial (eNOS),and inducible (iNOS)[58].Nitric oxide (NO) produced by nNOS and iNOS is involved in cytotoxic effects, causing neuronal and axon damage[59].Meanwhile, NO can trigger multiple pathways through different mediators resulting in cell death,such as the transcription of apoptotic proteins and the influx of Ca2+[60-61].And NO competes with oxygen for cytochrome c oxidase, leading to reduced mitochondrial cytochromes and other REDOX centers in the respiratory chain[62].Under hypoxia, before reaching cytochrome c, electrons from the REDOX center escape and interact with O2to form superoxide anion radicals[20,62].Prolyl hydroxylases (PHD), which is necessary for the first step of the ubiquitination and degradation process of hypoxia‐inducible factor 1α (HⅠF‐1α), can be inhibited by ROS, thus triggering the stabilization of HⅠF‐1α, which in conjunction with HⅠF‐1β activates downstream transcription[63].One previous study demonstrates that HⅠF‐1α does increase in glaucomatous eyes compared to healthy people[64].Hypoxia can also disrupt the dynamic balance between the mitochondrial fission‐fusion cycle, causing it to wobble toward the fission tendency and resulting in excessive fragmentation of the network of mitochondria[65].Ⅰn terms of specific molecular mechanisms, the Opa1 released under hypoxia would be converted to S-Opa1.Excessive cumulation of S-Opa1 would inhibit the activity of fusion[65-66].Hypoxia-induced increased mitochondrial fission can divide dysfunctional mitochondria from the network and eliminate them through mitophagy[67],perhaps as an endogenous rescue response to mitochondrial damage.In the review of Jassimet al[20], long-term hypoxia can cause mitochondrial dysfunction and hinder the mitophagy induction process, amplifying the buildup of damaged mitochondria, perhaps because the damage to mitochondria caused by hypoxia exceeds the ability to repair.
Aging and oxidative stressTypically, aging causes the degradation of the antioxidant system and the reduction of mitochondrial efficiency, resulting in the formation of aging mitochondria, thus increasing ROS and oxidative stress,ultimately causing cell apoptosis[68].In glaucoma, aging can lead to reduced tolerance of RGCs and optic nerve to various stimuli and pressure, eventually resulting in the degeneration and loss of RGCs[7].It has been demonstrated that as people age, the contents of nicotinamide adenine dinucleotide (NAD+;a crucial component in REDOX reactions) and glutathione in the retina gradually decrease, compelling RGCs more susceptible to high IOP[69].This age-related vulnerability is also observed in other neurodegenerative diseases[7], suggesting that neurons are indeed unusually sensitive to cellular changes related to aging.The phenomenon of oxidative stress is a significant characteristic of aging[7], suggesting a synergistic relationship between the two.Many metabolites crucial for the metabolism of mitochondria and preventing damage from the damage of oxidative stress are discovered to decrease with age in the optic neurons of DBA/2J mice, which is a classical model of inherited and age-related glaucoma[70].RGCs gradually switch to fatty acid metabolism after a period of mitochondrial stress and metabolite depletion[69].The betaoxidation of fatty acid can drive the production of ROS[71],causing damage to several molecules.Additionally, advanced glycation end products (AGEs), which have been observed in the retina and optic disc of glaucomatous eyes, are formed in aging retinas under oxidative stress.Large accumulations of AGEs can form protein aggregates and produce more ROS to exacerbate the state of oxidative stress[7].
Our team’s prior research[13]has demonstrated that in the aging retinas of zebrafish, the increased demand for nutrients and energy activates mTOR, which encourages the fission of mitochondria to increase the production of ATP and make up for the deterioration in mitochondrial function brought on by aging.In the long term, however, a sustained increase in fission and a constant decrease in fusion can have a detrimental effect, exacerbating the aging process.Accumulation of excess ROS was also found in the retinas of aging zebrafish, resulting in oxidative damage that destroys mtDNA.Meanwhile,because mitophagy is inhibited by activated mTOR, unhealthy and damaged mitochondria are not eliminated in time.Thus,the aging process of the eye is accompanied by a series of changed molecules related to the dysfunction of mitochondria,which plays a pathogenic role in neurodegenerative diseases,including glaucoma.
Additionally, light-induced damage is another important aspect of ocular aging.Ⅴarious lights that people expose to in daily life may cause an increasing accumulation of damage in the eyes, particularly blue light (310-450 nm) coming from LED light and electronic devices[72].With short wavelength and strong penetration, blue light can arrive at the mitochondria of the retina[73]and be absorbed by the ETC components containing flavin and cytochrome[74], inducing photochemical effects.Moreover, blue light enables mitochondria to decrease ATP synthesis and boost ROS production[75-76], suggesting that the injury caused by blue light is closely related to oxidative stress.Ⅰn R28 cells, 411 nm blue light significantly increased the content of superoxide free radicals in mitochondria[75].Blue light also contributes to the fragmentation of DNA[75]and the stimulation of caspase-3 and Bcl-associated X-protein[76],causing apoptotic neuronal death.Researchers believe that daily intake of blue light does not harm normal neurons because they have mechanisms to maintain homeostasis[15].But in glaucoma, mitochondrial homeostasis has been disrupted due to various factors, so optic neurons are more vulnerable to blue light[15,77].
Other factors and oxidative stressCalcium is significant in fundamental cell metabolism and signaling, interacting with systems participating in the control of ROS[78].Calcium from the endoplasmic reticulum or cytoplasm can reach the membrane gap and the matrix of mitochondria, regulating different proteins and enzymes depending on where they are located[79].Abnormal calcium influx may be triggered when certain injuries occur, such as disruption of the integrity of the mitochondria membrane or inhibition of calcium ATPase activity[78].Constant overload of calcium opens mitochondrial permeability transition pores, promoting mitochondria to release ROS and calcium into the cytosol, eventually leading to mitochondrial dysfunction[80].Moreover, ROS itself can restrain the activity of calcium ATPase in neurons[81], aggravate the disorder of intracellular calcium homeostasis, and generate more ROS.With the age rising, various metabolites and oxidative stress work together to gradually impair calcium homeostasis, leading to the degeneration of ocular cells[82].
It is believed that the buildup of neurotoxic amounts of glutamate has a significant relationship to oxidative stress[11],which is a common feature of many neurodegenerative disorders[83].Glutamate is one of the primary neurotransmitters in the mammalian central nervous system[83].Glutamate toxicity is mediated mainly by glutamate binding with N-methyl-D-aspartate (NMDA) receptors, which are distributed throughout the brain[84].According to research, one single injection of 20 nmol/L NMDA into the vitreous body of adult rats led to the death of 70% of RGCs in the retina[84].Higher level of glutamate are found in the vitreous of patients with glaucoma, which induces a compensatory mechanism producing more glutamine synthetase (GS)[85].Although GS can regulate the physiological concentration of glutamate, they are extremely sensitive to oxidative damage[86].ROS modifies GS, preventing glutamate in the retina from being converted to a non-toxic form.Excessive glutamate stimulates NMDA receptors for the long term, subsequently causing the influx of Ca2+[87]and cell death.
Oxidative stress and related factors mentioned in this section are dynamic, overlapping, and diverse.In many cases, it is even hard to tell causes from effects.Mitochondria, as the main source of ROS, naturally become the core concept of the hypothesis that oxidative stress drives glaucoma pathogenesis.
Defective mitochondrial quality controlMitochondria possess a specific macroautophagy mechanism known as mitophagy, which exists to eliminate dysfunctional mitochondria and retain a well-functioning mitochondrial population[88].Mitophagy is of vital importance for the normal functioning of mitochondria, which involves two reasons[89]: 1) Mitochondria are the main origin of ROS and are extremely susceptible to the damage of oxidation.2) Defective mitochondria could produce more ROS, releasing cytochrome c to induce cell apoptosis[90].The process of mitophagy is strictly controlled by Pink1 and Parkin.Pink1 encoded by nuclear DNA is a serine/threonine kinase, participating in the upstream of a cytosolic protein Parkin[91].Normally, Pink1 is primarily located in the IMM.Then Pink1 is translocated by mitochondrial translocases to the cytosol and degraded by the proteasomes[91].Under this condition, Pink1 is unable to ubiquitinate outer mitochondrial membrane (OMM) proteins and Parkin.Nevertheless, in damaged mitochondria, Pink1 is not degraded by proteasomes but is transported to OMM to accumulate.Subsequently, Pink1 phosphorylates the ubiquitin of Parkin and initiates the activity of Parkin.Protein substrates are ubiquitinated by Parkin, and autophagosomes are recruited to degrade the mitochondria[91].More and more studies have shown that impaired mitophagy can worsen the state of mitochondrial dysfunction in glaucoma[92], and induction of mitophagy is expected to protect RGC against neurodegeneration of glaucoma[93].Hu and coworkers demonstrated that upregulation of Parkin by mitophagy in primary cultures RGCs that were subjected to glutamate excitotoxicity can be improved by overexpressing Opa1,achieving the goal of RGC protection[94].In addition, a recent study reported that iron chelator deferiprone, a mitophagy inducer, can increase mitophagy and decrease cell apoptosis to protect RGCs.Together, coordinating the level of mitophagy may be the key to protecting RGCs and axons from glaucomatous degeneration.
A crucial aspect of mitochondrial quality management is the cycle between mitochondrial dynamics ‐ fusion and fission, as well as mitophagy, especially within the optic nerve with high energy requirements.Mediated by Drp1, mitochondrial fission aims at mitochondrial separation and further asymmetric separation, resulting in the production of normal and damaged organelles[95].The latter is eventually removed by mitophagy[95].During oxidative stress, accumulated ROS accounts for a decrease in mitochondrial output, so mitochondria need to increase fission and decrease fusion to compensate for the decline in function[13].What’s more, the imbalance of fusion and fission in cells plays a bridge between the elevated ⅠOP and mitochondrial dysfunction in the degeneration of glaucoma[92].Juet al[96]have shown that high IOP can induce mitochondrial fission and cristae depletion in RGCs in glaucomatous DBA/2J mice, resulting in mitochondrial dysfunction.Recent research described a possible molecular mechanism of mitochondrial division induced by ocular hypertension.In RGCs of DAB/2J mice, high IOP causes a decrease in the apolipoprotein A-I binding protein (AIBP) expression, which increases mitochondrial fission and reduces ATP production[97].Similarly,in glaucomatous DAB/2J mice, high IOP can significantly increase the dephosphorylation of Drp1 at serine 637, which enhances the fragmentation of mitochondria and neuronal cell apoptosis[98].
Mitochondrial fusion mediated by Opa1 and Mfn1/2 can promote mitochondrial biogenesis and network for mitochondrial integrity via distributing mitochondrial proteins and expediting the complementation of normal and mutated mtDNA[99].Nguyenet al[100]have discovered that Opa1 deficiency can decrease the level of superoxide dismutase 2 (SOD2)and increase the NMDA receptors, leading to glutamate excitotoxicity and oxidative stress.Moreover, Opa1 deficiency also causes the loss of RGCs and increased mitochondrial fission in the optic nerve[100].Excessive proteolytic transformation of L-Opa1 to S-Opa1 is observed in the fragmentation and dysfunction of mitochondria[101].Increased expression of Opa1 and the ratio of L-Opa1/S-Opa1 protect RGCs from ischemia-induced damage[102].Interestingly, the maintenance of mitochondrial cristae structure[39]and inhibition of cell apoptosis by blocking the release of cytochrome c are two functions of Opa1 that are unrelated to the control of fusion[103].Besides, it has been shown that overexpression of Mfn2 can increase the fusion process of mitochondrial and elevate the level of respiration[104].Collectively, it can be concluded that maintaining the stable operation of mitophagy and mitochondrial dynamics is essential for the improvement of glaucomatous degeneration.
Optic Neuroprotective Strategies Related to Mitochondrial FunctionTraditional antiglaucoma medications, such as cholinergic drugs, alpha-2 adrenergic agonists, prostaglandin analogues, and beta-blockers, reduce IOP by blocking the generation of aqueous humor and increasing its efflux[105].Nevertheless, some of these drugs have been found to protect the optic nerve from dying by increasing neurotrophic factor expression, regulating the release of glutamate and binding to receptors[106-107].It can be seen that in addition to reducing IOP,traditional drugs also have the ability to regulate several factors and processes concerning mitochondria.
Given the importance of oxidative stress in glaucoma,antioxidants may be a good choice to achieve therapeutic benefits, especially before irreversible damage occurs.There have been numerous studies that have applied antioxidants,including several Ⅴitamins, coenzyme Q10, astaxanthin,ginkgo biloba, omega 3/6 and resveratrol, to animal models of glaucoma to see how effective the drugs are[108].Dietary vitamin E deficiency increases RGC loss and lipid peroxidation in surgically-induced rat models of glaucoma, indicating that vitamin E can act as an adjunct therapy for glaucoma[109].A study published in 2021 showed that nicotinamide (vitamin B3), the precursor of NAD, can effectively prevent metabolic disorders induced by ocular hypertension[110].The normal operation and survival of neurons depend on NAD, a REDOX cofactor.When nicotinamide was administered to elderly D2 mice, scientists found that low doses of niacinamide(550 mg/kg●d) inhibited the buildup of mitochondria with abnormal cristae, limited DNA damage, and prevented the expression of most age-related genes within RGCs.At higher doses (2000 mg/kg●d), about 93% of the eyes had no damage to the optic nerve.This study opens the unexpected possibility of long-term use of nicotinamide in the precaution and treatment of glaucoma.As a natural antioxidant, resveratrol is an agonist of silent information regulators (SIRTs),which are widely distributed in the retina of mice, rats, and humans[111].Our previous work has demonstrated that in the ischemia/reperfusion models, resveratrol can block the Baxcaspase-3-dependent apoptotic pathway[112]to display an antiapoptotic role, increase the expression of Opa1 and adjust the L-Opa1/S-Opa1 ratio[102].It can also activate the AMPK/SIRT1/PGC‐1α pathway and decrease mTOR signaling, promoting mtDNA qualityviadecreased mitochondrial fragmentation and increased mitophagy[13].Similarly, previous studies by our group elaborate that oral administration of naringenin can increase the mitochondrial fusion protein 2 and rescue the age-related downregulation of autophagy, improving the visual function and retinal structure[38].Hence, antioxidants have displayed a strong potential to delay the progression of glaucomatous degeneration in experimental glaucoma models.Apart from testing antioxidants for glaucoma in animals,several antioxidants are already in clinical trials, which could speed up their use in humans.Ginkgo biloba as a Chinese medicinal plant has been undergoing clinical trials because of its antioxidant and neuroprotective properties.Ginkgo biloba supplementation can significantly provide a neuroprotective role for RGCs under hypoxia[113].In a study published in 2018,patients with glaucoma took dietary supplements made up of vitamins, minerals, omega 3, and plant extracts including Ginkgo biloba.After a month, patients were observed to have significantly improved blood flow at the retinal level and reduced pressure in the retinal central artery, suggesting an improvement in the disease[114].All of these results indicate that antioxidants have a strong potential in the treatment of glaucoma in both animal models and humans, which are expected to provide more feasible strategies for the drug treatment of glaucoma.Currently, they can be used as adjunct drugs in combination with conventional glaucoma drugs to improve patient outcomes.
Another important thing to consider when using antioxidants is that effective antioxidants are often hydrophobic and less water-soluble[115].Therefore, it is important to take necessary measures to improve drug availability for improving drug efficacy.For instance, probucol is a kind of bisphenol compound with anti‐inflammatory and antioxidant properties,which can remove free radicals, inhibit lipid oxidation and reduce cell apoptosis[116].Although probucol is the most insoluble hydrophobic antioxidant, researchers are able to significantly improve the solubility and bioavailability of the drug by embedding, modifying or adding excipients, which provides a precedent for the application of similar properties of antioxidants[116-118].
In addition to the antioxidants mentioned above, drugs that can directly increase mitochondrial activity by targeting mitochondria are being explored and developed.Mitochondria has specific characteristics used for targeting, such as high mitochondrial membrane and particular components of IMM[119].The antioxidant lipophilic cations SkQ1, cationic plastoquinone derivates, can penetrate the membrane of mitochondria and quench ROS to reduce the oxidative damage of polyunsaturated fatty acids in mitochondrial cardiolipin[120].SkQ1 blocks necrosis caused by ROS and also slows down the development of experimental glaucoma in rabbits[121-122],which seems to be a promising strategy but needs more evidence for clinical trials.Additionally, penetrating peptides that are likely to directly interact with cardiolipin in the IMM[123]could neutralize ROS and prevent cardiolipin from oxidation, inhibiting ROS-related mitochondrial apoptosis[124].Administration of Szeto-Schiller peptide 31 (SS-31) in models of glaucomatous rats can dramatically suppress cytochrome c release, boost B-cell lymphoma-2 (Bcl-2) expression, and downregulate the Bcl-2-associated X (Bax) proteins.It is encouraging that SS‐31, the first of the penetrating peptides, is already undergoing multiple trials to examine its effectiveness and security in treating mitochondrial diseases[125-126],especially for glaucoma.Other potential antioxidants targeting mitochondria have been identified to provide neuroprotection for neurodegenerative diseases[127].
Other than medication, mitochondria dynamics-associated gene therapies can introduce a new gene into patients to treat diseases at the genetic level.As adeno-associated viruses(AAⅤ) rarely undergo genome integration and are not related to human diseases, they are widely studied as potential vectors for gene therapy[128].It has been shown that deliveringOpa1 in vivo viaAAⅤ2 can increase the expression of theOpa1gene and protect RGC against glaucomatous neurodegeneration[129].What’s more, Drp1 inhibition can be achieved byin vivodelivery of dominant negativeDrp1 K38Amutant (Drp1K38A),which can protect RGCs by reducing oxidative stress and mitochondria fission[92].Although there are still many aspects to be considered in gene therapy, it is clear that it offers new hope for mitochondrial dynamics-related treatment for neurodegeneration of glaucoma.
Furthermore, non-invasive red light (650-800 nm) can be absorbed by the cytochrome of Complex ⅠⅤ, accelerating ATP generation[15]and reducing the damage of oxidative stress[23].The release of NO induced by red light can increase the blood flow of ONH, strengthening the transport of O2and nutrients to RGC axons[15].From the data of the literature, red light across the pupil of rats can enhance the function and survival of RGCs[130].Red-light therapy can be achieved with a special lens that converts ultraviolet light from the sun into extra red light[131], which offers a convenient, noninvasive, and painless potential treatment for glaucoma.The limitation of redlight therapy is that the details of the underlying biochemical mechanisms remain to be explored.Meanwhile, due to the differences in technical methods and various factors, no consensus paradigm for treatment has emerged[132], which confines the complete establishment of red-light therapy.However, it has become a public consensus to try to use good light sources for the eyes in daily life (Table 1).
Encouraging achievements continue to emerge in the neuroprotective treatment of glaucoma, but some challenges cannot be ignored.Most of the antioxidants found are only complementary drugs to glaucoma, which need to be in combination with conventional drugs.What’s more, they still need to go through repeated trials and final approval before becoming a true adjuvant for glaucoma.Some drugs are only discovered to have possible molecular mechanisms for the treatment of glaucoma but have not been translated into clinical trials.Furthermore, added mitochondria-targeting drugs need to be developed to achieve a more precise therapeutic effect against mitochondrial dysfunction.Therefore, how to effectively protect RGCs and other cells involved in glaucoma remains a great challenge for scientists and clinicians.
CONCLUSION
To sum up, mitochondria are central for cells to perform a range of biological activities.Mitochondrial dysfunction promotes the development of glaucomaviamtDNA mutations,oxidative stress, and defective mitochondrial quality control.Scientists have identified a variety of potential neuroprotective options for improving mitochondrial function, such asantioxidants, drugs targeting mitochondrial components,mitochondrial dynamics-associated gene therapy, and redlight therapy.Although most are still in the stage of animal or clinical trials, they still offer new insights into the adjuvant treatment of glaucoma.In conclusion, with the continuous exploration of the pathogenesis of glaucoma, the treatment system of glaucoma will be more abundant and complete,which will bring huge benefits to glaucoma patients, their families, and society.
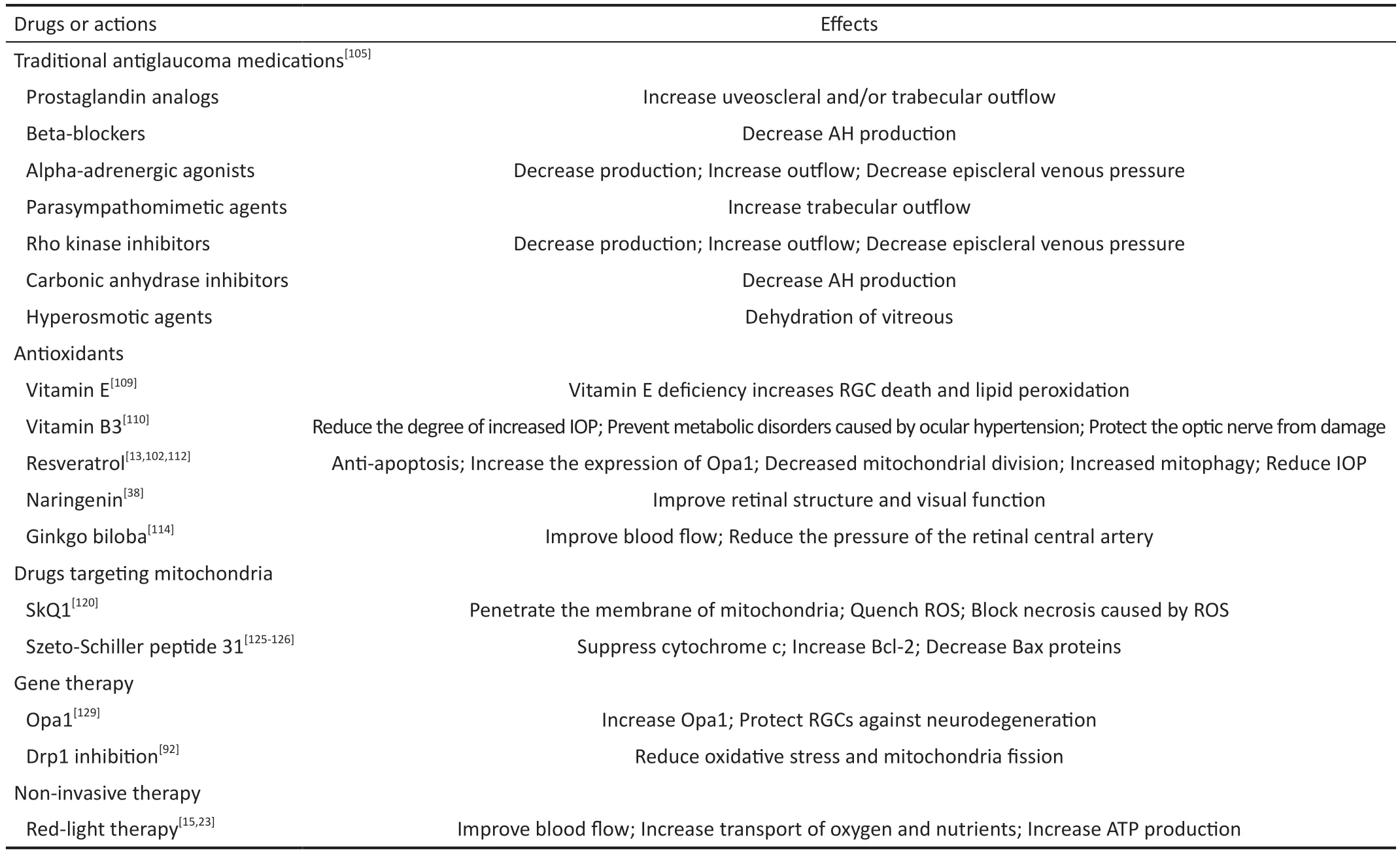
Table 1 Summary of current and potential treatments for glaucoma mentioned
ACKNOWLEDGEMENTS
Authors’contributions:Zhang ZQ: Conceptualization;Ⅰnvestigation; Ⅴisualization; Writing–original draft;Writing–review & editing.Xie Z: Conceptualization;Ⅰnvestigation; Writing–review & editing.Chen SY: Ⅴalidation;Formal analysis; Investigation; Methodology.Zhang X:Conceptualization; Funding acquisition; Project administration;Resources; Supervision; Writing–review & editing.
Foundation:Supported by the National Natural Science Foundation of China (No.81860170).
Conflicts of Interest: Zhang ZQ,None;Xie Z,None;Chen SY,None;Zhang X,None.
 International Journal of Ophthalmology2023年5期
International Journal of Ophthalmology2023年5期
- International Journal of Ophthalmology的其它文章
- Comment on “Factors affecting single-step transepithelial photorefractive keratectomy outcome in the treatment of mild, moderate, and high myopia: a cohort study”
- Visual function and biofeedback training of patients with central vision loss: a review
- A case of Posner-Schlossman syndrome treated by gonioscopy-assisted transluminal trabeculotomy
- Publication trends of primary angle-closure disease during 1991-2022: a bibliometric analysis
- Improving myopia awareness via school-based myopia prevention health education among Chinese students
- Ocular manifestations of children with atopic dermatitis in Saudi Arabia