
基于加权基因共表达网络分析方法筛选并鉴定结直肠癌的驱动基因
2023-05-12 07:43张和平王敬敏
新乡医学院学报 2023年5期
张和平,王敬敏,任 萍
(1.焦作市人民医院肛肠科,河南 焦作 454000;2.焦作市人民医院不孕不育门诊,河南 焦作 454000;3.焦作市人民医院耳鼻喉科,河南 焦作 454000)
结直肠癌(colorectal cancer,CRC)是世界范围内常见的消化系统恶性肿瘤。随着经济的发展、饮食习惯和生活方式的改变,CRC发病率和病死率逐年上升,且发病呈年轻化趋势[1]。CRC的发生是一个复杂的多基因、多步骤、多阶段过程,常常多个基因相互作用,包括癌基因激活和抑癌基因失活[2]。因此,寻找调控CRC发生发展的核心基因和有效的治疗靶点是当前研究的重点。本研究使用WGCNA方法,基于148个人类CRC样本的2 000个基因组成的数据集构建共表达网络,分析各模块与CRC临床特征的相关性,寻找与肿瘤进展高度相关的基因,以期为CRC的临床治疗提供靶点。
1 资料与方法
1.1 细胞、主要试剂和仪器CRC细胞株LOVO、SW620、HCT116、SW480、RKO、CaCo2和对照肠黏膜上皮细胞NCM460均来自ATCC细胞库。胎牛血清购自美国Gibco公司,TRIzol试剂、反转录试剂盒、SYBR Green I购自日本TaKaRa公司,细胞计数试剂盒-8(cell counting kit-8,CCK-8)购自江苏凯基生物技术有限公司,CYTH1、甘油醛-3-磷酸脱氢酶(glyceraldehyde-3-phosphate dehydrogenase,GAPDH)引物序列信息来自Primer Bank数据库,si-NC、si-CYTH1干扰片段购自苏州吉玛基因有限公司;细胞培养箱购自美国Thermo Fisher Scientific公司,酶标仪UMR9600购自杭州优米仪器有限公司,Transwell过滤器购美国康宁公司。
1.2 数据来源与CRC相关的数据来自NCBI基因表达综合数据库(gene expression omnibus,GEO) (http://www.ncbi.nlm.nih.gov/geo),数据集为GSE21510,共148个样本,芯片平台为GPL570。使用GEO2R工具对差异基因进行分析,根据P值和基因表达fold-change对差异基因进行排序[3-4],选择前2 000个样本进行进一步分析。
1.3 CRC共表达模块的分析与构建使用下载的CRC芯片基因表达数据构建无标度网络,用加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)算法继续进行模块构建,在R包(http:/www.r-project.org/)中运行WGCNA,分析与临床性状之间的交互强度[5-6]。估算出模块与临床特征的相关性,鉴定出与临床表型高度相关的模块[7-8];以P<0.05的模块为显著相关的模块。
1.4 Cytoscape分析模块的核心基因提取与临床特征相关性最高的模块内基因,将基因导入Cytoscape,分析核心基因,用MCODE插件筛选核心蛋白[9]。选择与其他基因连接最多的枢纽基因CYTH1为核心基因。
1.5 细胞培养将LOVO、SW620、HCT116、SW480、RKO、CaCo2和NCM460细胞接种于含 RPMI 1640 (Hyclone)的培养基中,添加胎牛血清(fetal bovine serum,FBS),置于37 ℃、含体积分数5% CO2培养箱中培养,取对数生长期细胞用于后续实验。
1.6 实时荧光定量-聚合酶链式反应 (quantitative real-time polymerase chain reaction,RT-qPCR)法检测细胞中CYTH1表达取对数生长期LOVO、SW620、HCT116、SW480、RKO、CaCo2和NCM460细胞,使用TRIzol试剂提取总RNA;利用反转录试剂盒将RNA反转录为cDNA。用SYBR Green I对样品进行qRT-PCR分析。CYTH1正向引物序列为5′-GACGACAGCTACGTTCCCAG-3′,反向引物序列为5′-TCCTGTTCTCCGTCGGATG-3′,扩增产物大小为140 bp。内参为GAPDH[10],GAPDH正向引物序列为5′-GACTCATGACCACAGTCCATGC-3′,反向引物序列为5′-AGAGGCAGGGATGATGTTCTG-3′。反应体系:TB-Green 10.0 μL,灭菌蒸馏水 6.8 μL,cDNA溶液 2.0 μL,上下游引物各0.4 μL,ROX Reference Dye 0.4 μL;反应条件为:95 ℃预变性1 min,95 ℃变性15 s,60 ℃退火15 s,72 ℃延伸34 s,共40循环,检测各样品的Ct值,采用2-ΔΔCt法计算CYTH1在各细胞中的相对表达量。应用GraphPad Prism 6软件进行图形构建[11]。
1.7 CCK-8法检测细胞增殖能力取对数生长期HCT116和SW480细胞,分别转染si-NC、si-CYTH1干扰片段,转染基因干扰片段后24 h以每孔1 000个细胞接种于96孔培养板中进行培养,每孔100 μL,每孔加入10 μL CCK-8试剂,37 ℃继续孵育2 h。在酶标仪上于450 nm波长处测定每孔的吸光度(absorbance,A) 值,连续监测4 d,根据数值绘制细胞增殖曲线,并计算细胞增殖率。实验重复3次,取均值。
1.8 Transwell实验检测细胞迁移率取对数生长期HCT116和SW480细胞,分别转染si-NC、si-CYTH1干扰片段,24 h后将si-NC-HCT116、si-CYTH1-HCT116、si-NC-SW480、si-CYTH1-SW480细胞用胰酶消化后,反复吹打细胞悬液,使细胞充分分散,于显微镜下计数细胞数量。细胞计数后每个小室接种 250 μL无血清培养基稀释的细胞悬液,每个小室中(1~5)×105个细胞,下室加入600 μL含体积分数20%血清的1640 培养基;培养24 h后,在显微镜下(×200)观察穿过微孔进入下室的细胞,计算迁移细胞数。

2 结果
2.1 CRC数据集的预处理结果使用GEO2R软年对差异表达基因进行分析,发现45 198个基因的P值均大于0.05。根据P值对差异基因进行排序,使用flashClust函数对前2 000个基因进行聚类分析。对样本进行聚类以检测离群值,所有样本都在聚类中,白色为低,红色为高。结果显示,148个样本产生了2个主簇,其中正常组织有25个样本,肿瘤组织有123个样本(图1)。

图1 CRC相关样本的聚类分析Fig.1 Cluster analysis of samples associated to CRC
2.2 共表达模块应用WGCNA方法对148个样本的 2 000 个基因数据集进行分析。构建WGCNA时,选择软阈值来进行共表达相邻基因相似度筛选。基于无标度拓扑准则,根据拟合指数和网络平均连接程度,确定相关系数接近0.90时,最佳软阈值β=12,并构建基因网络(图2)。
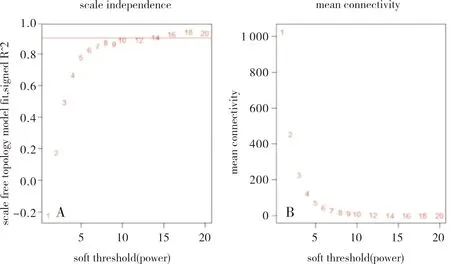
A:soft threshold (power)表示权重,纵坐标表示无标度拓扑模型拟合;B:soft threshold (power)表示权重,纵坐标表示平均连接度。图2 共表达模块软阈值的确定Fig.2 Determination of soft threshold in coexpression module
2.3 各模块与临床特征的相关性结果见图3。WGCNA构建的模块用不同颜色表示,不属于任何模块的基因被放置在灰色模块中。灰色基因模块在本研究中被忽略。计算基因表达模块的关联,中间的黄色部分表示模块之间的相关性。横轴和纵轴的不同颜色代表不同的模块。浅颜色表示拓扑重叠小,深颜色表示拓扑重叠大。分析模块与临床特征的相关性,每一行对应一个模块特征基因,列对应一个临床特征。每个单元格包含相应的相关性和P值。结果显示,turquoise模块与CRC临床转移特征显著相关。绘制特征基因与临床性状相关的散点图,发现turquoise模块核心基因与肿瘤转移呈明显相关性,蓝色模块基因与肿瘤转移无明显相关性。

A:CRC共表达基因聚类树型图;B:基因表达模块的关联;C:模块与临床特征的关联;D:蓝色模块特征基因与肿瘤转移的相关性;E:Turquoise模块特征基因与肿瘤转移的相关性。图3 各模块与临床性状的相关性Fig.3 Correlation of each module and clinical characteristics
2.4 核心基因筛选结果见图4。将turquoise模块基因及其相关文件导入Cytoscape软件,分析核心基因。因为第一个模块包含的基因太多,超过1 000条边,所以只显示关键基因,黄色为核心基因。Cytoscape 软件识别出了排名前3位的核心模块,3个模块中的核心基因分别为CYTH1、LOC157273和RP11-744D14.2,其中,与其他基因连接最多的枢纽基因是CYTH1。
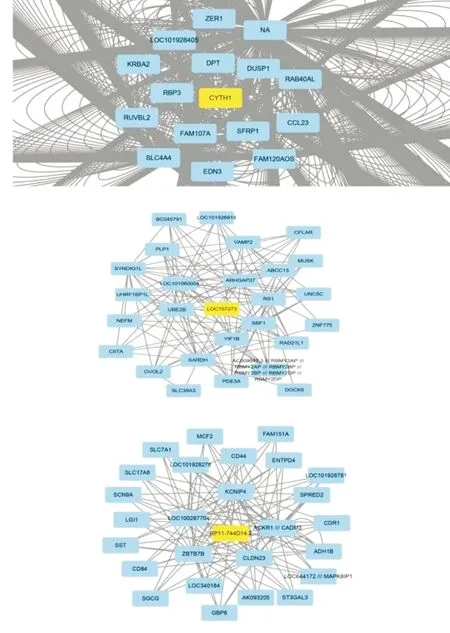
图4 前3位的核心模块中核心基因的可视化Fig.4 Visualization of core genes in the top three core modules
2.5 CYTH1 mRNA在结直肠癌组织与癌旁组织、对照结直肠组织及7种细胞中的表达水平比较基于Oncomine数据库分析结果显示,与癌旁组织相比,CRC组织中CYTH1 mRNA表达水平显著下调;与对照结直肠组织标本相比,CRC标本中CYTH1表达下调约75%(图5)。RT-qPCR 检测结果显示,LOVO、SW620、HCT116、SW480、RKO、CaCo2和NCM460细胞中CYTH1 mRNA相对表达量分别为0.104±0.050、0.185±0.066、0.261±0.024、0.544±0.255、0.227±0.011、0.084±0.001、0.999±0.003。LOVO、SW620、HCT116、SW480、RKO和CaCo2细胞中CYTH1 mRNA相对表达量显著低于NCM460,差异有统计学意义(t=31.080、21.262、51.963、3.093、114.344、216.340,P<0.001)。

红色框表示表达上调,蓝色表示下调。图5 基于Oncomine数据库CYTH1 mRNA在结直肠癌组织和对照组织中的表达水平Fig.5 Expression levels of CYTH1 mRNA in colorectal cancer tissues and control tissues based on Oncomine database
2.6 CYTH1对HCT116和SW480细胞增殖和迁移能力的影响结果见图6。干预1、2、3、4 d,si-NC-HCT116细胞增殖率分别为-(4.44±18.46)%、(258.33±92.04)%、(51.76±29.17)%、(49.66±22.17)%,si-CYTH1-HCT116细胞增殖率分别为-(13.77±36.64)%、(317.86±151.72)%、(69.59±20.14)%、(33.65±3.09)%;不同时间点,si-NC-HCT116细胞与si-CYTH1-HCT116增殖率比较差异无统计学意义(t= 0.321、-0.474、-0.711、1.011,P>0.05)。si-NC-SW480细胞增殖率分别为-(0.36±3.80)%、(488.89±164.05)%、 (124.17±27.41)%、(43.18±18.97)%,si-CYTH1-SW480细胞增殖率分别为-(0.23±4.23)%、(550.00±298.45)%、(123.00±31.11)%、(40.90±8.00)%;不同时间点,si-NC-SW480 细胞与si-CYTH1-SW480细胞增殖率比较差异无统计学意义(t= 0.148、-0.254、0.040、0.157,P>0.05)。培养24 h后,si-NC-HCT116、si-CYTH1-HCT116 迁移细胞数分别为 24.60±2.65、86.80±6.68,si-CYTH1-HCT116迁移细胞数显著高于si-NC-HCT116细胞,差异有统计学意义(t=-17.318,P<0.001);si-NC-SW480、si-CYTH1-SW480 细胞迁移率分别为53.20±4.96、80.80±4.96,si-CYTH1-SW480 迁移细胞数显著高于si-NC-SW480细胞,差异有统计学意义(t=-7.876,P<0.001)。
3 讨论
WGCNA是一种用于复杂样本[12]的分析方法,利用高通量测序数据获取模块信息。在这种方法中,一个模块包括一组具有相似表达谱的基因。如果某些基因在生理过程中或在不同的组织中始终有类似的表达变化,那么这些基因是功能相关的,然后将它们定义为一个模块。当定义了一个基因模块后,可以利用模块与临床特征的相关性结果,找到最相关的基因模块,重点分析其中的基因[13]。WGCNA 可以描述不同样本之间的基因关联模式,可用于识别高度相关的基因集[14-15]。聚类分析仅仅考虑了单个基因之间表达模式相似度,而忽略了多个基因间的表达相关性,会损失一部分信息[16-17]。与只关注差异表达的基因相比,WGCNA利用变化最大的基因信息来识别感兴趣的基因,并与表型进行关联分析[18]。本研究使用WGCNA方法分析基因表达数据,由于输入基因数量有限,选择了 2 000 个差异最大的基因,并用WGCNA对NCBI获得的148个样本的2 000个不同表达基因进行了研究,最终确定了3个基因模块。
恶性肿瘤的主要生物学特征是其具有侵袭和转移的能力,这是肿瘤患者复发和死亡的主要原因。CRC的转移过程是动态的,其发生机制非常复杂。为了分析CRC转移的相关基因集,本研究使用WGCNA方法将获得的模块与临床特征联系起来,结果发现,turquoise模块与转移关系最为密切;将turquoise模块基因导入Cytoscape进行分析,得到3个核心基因CYTH1、LOC157273和 RP11-744D14.2,其中,与其他基因连接最多的枢纽基因是CYTH1。CYTH1是细胞黏附蛋白家族成员,在自然杀伤细胞和外周血T细胞中高表达,调控整合素在淋巴细胞细胞膜上的黏附[19-20]。该家族成员具有相同的结构,包括N端卷曲螺旋基序、中央的Sec7结构域和C端PH结构域[21]。本研究基于 Oncomine 数据库分析了CRC细胞中CYTH1的mRNA表达水平,结果显示,CRC组织中CYTH1 mRNA表达水平显著低于癌旁组织, CRC组织中CYTH1表达水平较对照组结直肠组织下调75%。另外,本研究结果显示,SW620、HCT116、SW480、RKO和CaCo2细胞中CYTH1相对表达量显著低于NCM460;说明CYTH1在CRC中有可能是抑癌基因。
本研究进一步检测CYTH1对CRC细胞增殖和迁移的影响,结果显示,不同时间点,si-NC-HCT116细胞与si-CYTH1-HCT116增殖率比较差异无统计学意义,且si-NC-SW480细胞与si-CYTH1-SW480增殖率比较差异亦无统计学意义,说明CYTH1对CRC细胞的增殖无明显影响。另外,本研究结果显示,培养24 h后,si-CYTH1-HCT116细胞迁移率显著高于si-NC-HCT116,且si-CYTH1-SW480细胞迁移率显著高于si-NC-SW480细胞说明 CYTH1可抑制CRC细胞的迁移能力。因此,推测CYTH1可能参与了CRC的进展过程,但具体机制尚不明确,需要进一步的研究。
综上所述,通过WGCNA分析找到与CRC细胞迁移相关的核心基因CYTH1,CYTH1在CRC组织和细胞中表达下调,CYTH1可抑制CRC细胞的迁移能力;因此,推测CYTH1在结直肠癌中起着抑癌作用,且有可能成为CRC有效的治疗靶点。
猜你喜欢
中学生数理化·高一版(2021年2期)2021-03-19
昆明医科大学学报(2020年12期)2021-01-26
河北渔业(2018年7期)2018-09-15
知识经济·中国直销(2018年8期)2018-08-23
健康必读·下旬刊(2018年6期)2018-07-24
山东医药(2017年20期)2017-07-01
数学学习与研究(2017年3期)2017-03-09
中国老区建设(2016年1期)2016-02-28
中国学术期刊文摘(2016年1期)2016-02-13
山东医药(2015年44期)2015-02-28
