
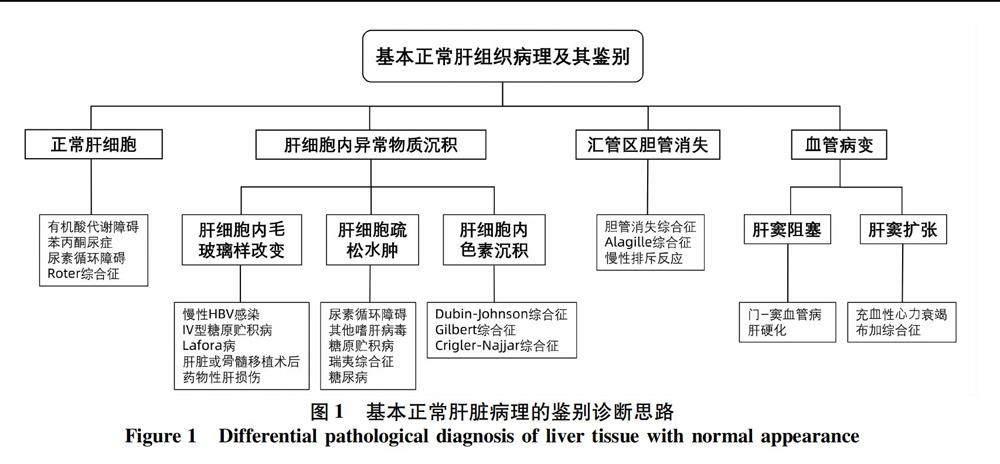
“基本正常肝组织”的病理鉴别诊断思路
2023-04-29 00:44:03张萌萌孟尧赵新颜
临床肝胆病杂志 2023年3期
张萌萌 孟尧 赵新颜
摘要:肝脏病理活检是明确肝脏疾病诊断的关键手段,对判断病情严重程度、决策治疗时机、预测疗效及预后具有重要价值。病理专家通过识别肝脏病理损伤模式判断病变性质,作为诊断及鉴别诊断依据,其中一类病理损伤模式,看似“基本正常”, 实则需要细致入微地观察易被忽略的病理变化,从而减少误诊或漏诊。本文重点介绍了 “基本正常”的肝组织病理诊断思路及鉴别诊断要点。
关键词:肝疾病; 病理学; 诊断, 鉴别
Differential pathological diagnosis of liver tissue with normal appearance
ZHANG Mengmeng, MENG Yao, ZHAO Xinyan. (Liver Research Center, Beijing Friendship Hospital, Capital Medical University & National Clinical Research Center for Digestive Diseases, Beijing 100050, China)
Corresponding author:
ZHAO Xinyan,zhao_xinyan@ccmu.edu.cn (ORCID:0000-0002-8016-4368)
Abstract:
Liver biopsy is a key method for clarifying the diagnosis of liver diseases and has an important value in determining disease severity, deciding treatment timing, and predicting treatment response and prognosis. By recognizing the pattern of liver pathological injury, pathologists evaluate the nature of lesions as the basis of diagnosis and differential diagnosis, and among these patterns, liver tissue with “normal appearance” requires careful observation of easily overlooked pathological changes, so as to reduce misdiagnosis or missed diagnosis. This article mainly introduces the thoughts in the pathological diagnosis of liver tissue with normal appearance and the key points in differential diagnosis.
Key words:
Liver Diseases; Pathology; Diagnosis, Differential
主要的肝臟病理损伤模式包括:汇管区炎症型、小叶内炎症型、急(慢)性胆汁淤积型、血管损伤型及肿瘤等[1]。此外,部分肝脏疾病肝组织活检低倍镜下观察,肝组织形态结构看似“基本正常”,多无明显的炎症坏死、脂变、细胆管反应及纤维化,需要在高倍镜下仔细观察才可以发现细微病理变化,以此为线索,结合临床进一步明确诊断。本文以肝脏病理读片顺序为主线,梳理了从肝细胞实质到汇管区间质可能发生的细微病变,以镜下肝脏病理特点为出发点总结相关需要鉴别的疾病。以发生率较高的疾病为主要论述对象,以与其肝脏病理特征类似的其他疾病为主要鉴别对象,旨在为镜下肝组织基本正常的肝脏疾病提供鉴别诊断思路(图1)。
1 完全正常肝组织形态结构
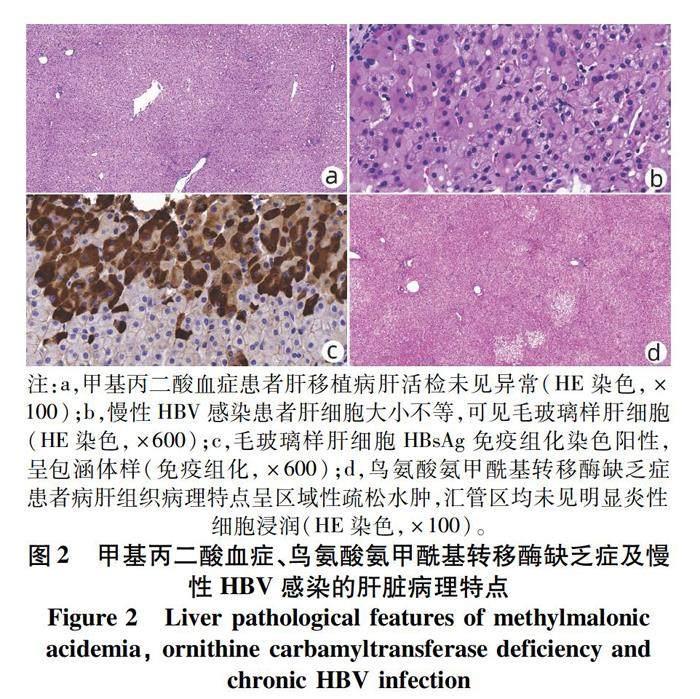
1.1 有机酸代谢障碍 有机酸代谢障碍又称有机酸血症或有机酸尿症,是由于某些酶的缺乏导致体内三大营养物质代谢过程中所产生的羧基酸及其代谢产物蓄积的一组疾病,属于遗传代谢性肝病[2]。甲基丙二酸血症和丙酸血症是常见的较易引起危重症的有机酸代谢障碍性疾病,为常染色体隐性遗传病。主要的临床特点是新生儿及婴儿期发生的急性酸中毒、高氨血症和中毒性脑病。肝脏病理为正常肝组织(图2a)。串联质谱检查测定血浆氨基酸水平、尿气相色谱质谱检测尿液有机酸水平可明确诊断。
1.2 鉴别诊断 对于组织学正常的肝脏疾病的诊断及鉴别诊断主要依靠临床表现、实验室检查、影像学检查、血尿代谢物筛查及基因检测。完全正常肝组织形态结构还可见于其他的遗传代谢病如苯丙酮尿症,部分尿素循环障碍以及Rotor综合征等。
苯丙酮尿症是一种常染色体隐性遗传病,由于苯丙氨酸羟化酶缺乏引起苯丙氨酸及苯丙酮酸堆积并从尿中排出。临床特点以生长发育迟缓及神经系统表现最为突出,可有鼠臭味、皮肤干燥及脑电异常[3]。光镜下肝组织表现正常[4]。患儿血浆苯丙氨酸明显升高,尿三氯化铁试验可协助诊断本病。
Rotor综合征是一种常染色体隐性遗传病,患者常婴幼儿期及青少年期发病,临床上主要表现为持续或间断黄疸,以非梗阻性高直接胆红素血症为主,有时可伴有间接胆红素升高,肝脏病理通常正常,诊断主要依靠基因检测,可发现SLCO1B1和SLCO1B3基因突变[5]。本病预后良好,早期诊断可避免过度治疗。
2 肝细胞内异常物质沉积
2.1 肝细胞内毛玻璃样改变
2.1.1 慢性HBV感染 毛玻璃样变是慢性HBV感染的特征性表现(图2b),由于HBsAg沉积于肝细胞内质网,镜下可见胞质内丰富的细颗粒物并呈轻度嗜酸性。有时胞质与增厚的胞膜之间可见清晰的光晕,细胞核常移位到肝细胞边缘[6]。毛玻璃细胞可成簇或成片排列。若病毒复制活跃,大量HBcAg累积可观察到肝细胞核呈磨砂状。HBsAg免疫组化染色可确认毛玻璃样变的性质(图2c)。
2.1.2 鉴别诊断 当HBsAg免疫组化染色为阴性,毛玻璃样改变还可出现在Ⅳ型糖原贮积病(支链淀粉样物质沉积)、Lafora病(异常糖原积累)、肝脏或骨髓移植术后以及药物性肝损伤的适应性反应。
由于Ⅳ型糖原贮积病肝细胞中贮积物为支链淀粉,即化学性质与淀粉类似的无分支多糖,肝细胞糖原染色(PAS染色)为阳性,由于支链淀粉不能被淀粉酶消化,淀粉酶消化后的肝细胞PAS染色仍为阳性。Lafora病毛玻璃细胞更倾向分布于汇管区周围,PAS染色阳性,淀粉酶消化后则为弱阳性[7],铁染色和甲基胺银染色均为弱阳性,HBsAg和HBcAg免疫组化染色均为阴性。单克隆抗体KM279染色可为阳性,具有特异性[8]。此外,苯巴比妥的应用也可导致肝细胞的毛玻璃样改变,即药物的适应性反应。
2.2 肝细胞疏松水肿
2.2.1 尿素循环障碍 尿素循环障碍是由于尿素合成途径中的代谢酶或转运蛋白的先天性或继发性缺陷或功能障碍引起体内氨的蓄积。其中以鸟氨酸氨甲酰基转移酶缺乏症和瓜氨酸血症最常见。此类疾病多发于婴幼儿,成年亦可发病,临床特点是由高氨血症引起的嗜睡、喂养不良、呕吐,病情进展可出现肝衰竭,颅压升高、肌张力降低、癫痫等神经系统症状,甚至死亡[9]。血氨及其代谢物筛查有助于诊断本病。适时行肝移植可改善大部分患者预后。在临床上,尿素循环障碍常无低血糖表现,同时阴离子间隙正常。反之则更倾向于有机酸代谢障碍[10]。
病理方面,肝脏的形态学变化可能更多地与患者肝活检时的状态有关。当患者处于疾病缓解期且血氨正常,镜下肝组织多无异常。当疾病进展到一定程度或处于发作期,镜下可见区域性肝细胞肿胀、胞质疏松水肿(图2d),有时可伴脂肪变性和汇管区间质纤维化[11]。对肝组织进行酶活性测定或基因检测有助于鉴别尿素循环障礙的不同类型。
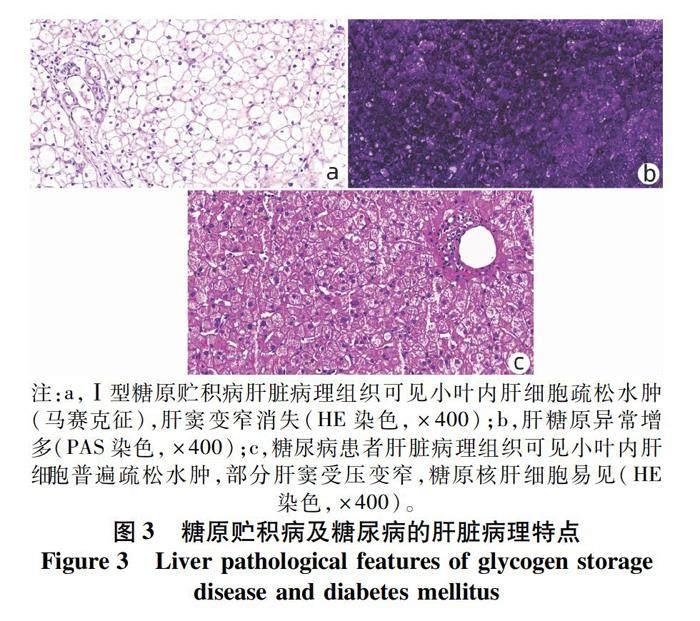
2.2.2 鉴别诊断 肝细胞肿胀还可见于糖原贮积病、瑞氏综合征、糖尿病等。糖原贮积病以Ⅰ型最常见,肝活检可见肝细胞肿胀,类似于植物细胞样外观,细胞膜增厚,由于糖原沉积致使细胞器向周围移位,呈马赛克样改变,PAS染色提示肝糖原异常增多(图3a、b)。胞质空泡状,可见大小不一的脂滴。电镜下可见糖原颗粒均匀分布于胞质和细胞器内[12]。瑞氏综合征患者肝活检可见细胞质疏松肿胀,肝细胞内有大量脂肪滴,脂肪染色阳性,电镜下可观察到肿胀的线粒体,患者常有病毒性感染及服用水杨酸类药物病史[13]。糖尿病患者由于胰岛素抵抗使肝细胞合成的糖原不能及时排出而在肝细胞蓄积,镜下可见细胞肿胀,疏松水肿,糖原核肝细胞易见(图3c),须根据患者既往病史进一步鉴别。肝细胞肿胀还可见于其他嗜肝病毒早期感染、EB病毒、巨细胞病毒感染相关肝炎等。
2.3 肝细胞内色素沉积
2.3.1 Dubin-Johnson综合征 Dubin-Johnson综合征是由于编码多药耐药相关蛋白2的ABCC2基因突变引起的一种常染色体隐性遗传疾病,基因突变导致胆红素等色素颗粒泵至毛细胆管发生障碍引起肝细胞黑色素颗粒蓄积[14]。临床特点主要是慢性黄疸,伴胆红素升高,以结合胆红素升高为主。
肝活检是诊断本病的金标准。镜下小叶结构基本完整,肝细胞大小正常,肝细胞内可见较典型的棕黑色色素颗粒,颗粒较粗大(图4a),聚积于中央静脉周围并向小叶周围延伸,Schmorl染色阳性(图4b),多数汇管区无扩大,无明显炎细胞浸润,无界面炎,铜、铁染色阴性。电镜下此种色素颗粒内可见致密的核心结构,可区别于其他胆红素升高的疾病。特别是Rotor综合征,该病肝细胞内无色素颗粒沉着[15],而临床上以直接胆红素升高为主要表现,SLCO1B1和SLCO1B3基因检测有助于本病确诊。
2.3.2 鉴别诊断 对于细胞内出现的大量棕黑色色素沉着,首先要鉴别铁颗粒沉积。铁颗粒沉积主要见于血色病。血色病早期肝活检仅在汇管区周围肝细胞中存在铁沉积,需仔细观察才能发现肝细胞中的折光颗粒(图4c、d)。随着疾病进展,可观察到汇管区周围肝细胞中大量含铁血黄素沉积,严重者可见小叶内弥漫性分布。 其次需要鉴别肝细胞内脂褐素颗粒沉积,主要见于Gilbert综合征和Crigler-Najjar综合征。此两种疾病均属常染色隐性遗传病,由于
尿苷二磷酸葡萄糖醛酸转移酶基因突变引起该酶活性减低或功能障碍,导致内质网内非结合胆红素转化为结合胆红素异常[16],临床上以间接胆红素升高为主。病理上,光镜下Gilbert综合征病理特点是肝细胞疏松水肿,可见均匀一致细小色素颗粒,多位于中央静脉周围(图4e)。Crigler-Najjar综合征与Gilbert综合征病理改变无明显差别,主要是肝细胞胞质内可见棕褐色胆色素颗粒沉积,通常无纤维组织增生及炎症坏死改变。Gilbert综合征和Crigler-Najjar综合征主要的鉴别点是基因突变的程度不同,前者苯巴比妥治疗有效[17]。
3 汇管区小胆管消失
3.1 胆管消失综合征(vanishing bile duct syndrome,VBDS) VBDS是指汇管区小胆管进行性破坏和消失。病因多样,大多数与药物有关,也可与免疫、感染及缺血缺氧等因素相关,少数属先天性胆管缺失。发病机制尚不明确,临床特点主要是皮肤瘙痒和黄疸,通常发生在重度胆汁淤积性肝炎发作后,部分伴有皮疹、发热和嗜酸性粒细胞增多。生化方面表现为血清碱性磷酸酶和胆红素持续升高。
肝活检是诊断本病的必要条件,肝活检发现小叶内胆管缺乏(活检标本至少有10个以上汇管区且胆管消失>50%)即可诊断本病[18](图5a),CK7/CK19染色有助于明确胆管消失程度(图5b)。不同原因引起的VBDS镜下可有不同表现,巨细胞病毒感染的病例还可在汇管区观察到炎性细胞浸润[19]。对于药物如抗生素引起的VBDS可见毛细胆管淤胆、汇管区炎症、纤维化以及肝细胞胆管化生。
3.2 鉴别诊断 Alagille综合征属常染色体显性遗传病,由于Notch通路异常(包括JAGGED1、NOTCH2)导致小胆管消失。临床主要表现为胆汁淤积导致的皮肤瘙痒和黄疸[20]。6个月及以上儿童肝活检典型镜下表现为明显的胆汁淤积和小叶间胆管减少,在汇管区周围肝细胞中可见铜沉积,假腺样肝细胞和蜡质样细胞,多不伴有小叶或汇管区炎症或细胆管反应。6个月以前患儿病理表现与新生儿胆道闭锁相似,以胆管增生、胆汁淤积和门静脉支扩张为主要表现[21]。提示本病的胆管消失是进行性发展。
胆管消失还可见于肝移植术后慢性排斥反应,肝活检可表现为VBDS,窦内可见炎症细胞浸润,还可见闭塞性动脉炎以及单个核细胞浸润[22],早期慢性排斥反应还可表现小叶中央坏死性炎症,晚期慢性排斥可见汇管区不同程度纤维化。
4 血管病变
4.1 门静脉支闭塞
4.1.1 门-窦血管病(porto - sinusoidal vascular disease, PSVD) PSVD是一种需要肝活检诊断的一种肝脏血管性疾病。发病机制尚不十分清楚,可能主要与遗传、药物毒物、自身免疫以及血液系统疾病有关。临床特点以门静脉高压为主,肝功能储备好,影像学检查提示门静脉、肝静脉和下腔静脉通畅。病理上以闭塞性门静脉病、结节性再生增生、不完全性间隔纤维化为主要特点(图6a、b)[23]。
4.1.2 鉴别诊断 PSVD主要与肝硬化鉴别。肝硬化在组织学上也存在结节再生,相比PSVD,肝硬化常可见完整的假小叶形成,肝硬化逆转时,纤维间隔变细、离断,可见汇管区残基,称为肝脏修复复合体(hepatic repair complex),易于与PSVD相混淆,患者既往肝硬化病史是与PSVD相鉴别的关键。
4.2 肝窦扩张
4.2.1 充血性心力衰竭 充血性心力衰竭是一种常见的临床疾病,会导致血液的淤滞和静脉系统回流压力增加;常见引起充血性心力衰竭的疾病有肺源性心脏病、缩窄性心包炎、风湿性心瓣膜病以及心肌病等。而心力衰竭引起淤血性肝病的发病机制主要与肝静脉压升高、肝血流量降低和动脉血氧饱和度降低有关[24]。临床表现有肝肿大、颈静脉怒张、腹水、凹陷性水肿、黄疸等;生化特点以碱性磷酸酶、γ-谷氨酰转肽酶及胆红素的升高为主,影像学检查可表现为下腔静脉及肝静脉的扩张。
由于下腔静脉及肝静脉淤血,肝脏病理镜下早期可见肝窦扩张伴淤血(图6c、d),有时可见早期靠近中央静脉的位于肝小叶Ⅲ区的血窦内皮细胞发生坏死和局灶性炎性细胞浸润,静脉内膜增厚引起小静脉管腔变窄导致肝窦微循环重新分布,肝细胞缺血缺氧,晚期星状细胞活化,纤维组织增生,可见窦周纤维化及中央静脉管壁纤维化。
4.2.2 鉴别诊断 布-加综合征(Budd-Chiari syndrome,BCS)又称巴德-基亚里综合征,定义为各种原因引起的从肝小静脉至下腔静脉回流至右心房入口的肝静脉流出道的梗阻或狭窄[25]。其发病机制主要包括先天性血管发育异常、血栓机化、机械性损伤等方面。目前BCS的主要诊断方法是影像学,可见肝静脉及肝段下腔静脉的阻塞。
BCS急性期可见小叶中心和中心区肝窦明显扩张淤血,肝静脉分支也可见有新鲜血栓,而汇管区多无明显病理改变[26];当BCS进入慢性期,中央静脉壁增厚,可见小叶中心-小叶中心的桥接纤维化,也可出现结节性再生性增生。
5 小结
综上,即使肝组织镜下所见基本正常,也需仔细观察肝脏病理的细微表现,识别肝组织病理形态学特点,如肝细胞内异常沉积物,汇管区门静脉支缺失或小胆管缺失等,以提高诊断的准确率、减少误诊及漏诊率。对于肝组织学诊断困难的肝脏疾病,需通过临床表现、生化、代谢筛查、影像学及基因检测综合分析以明确诊断并制订治疗方案。
利益冲突声明:
所有作者均声明不存在利益冲突。
作者贡献声明:张萌萌起草、撰寫文章;孟尧论文修改及支持性贡献;赵新颜对文章的知识性内容作批评性审阅,指导,支持性贡献。
参考文献:
[1]GASMI B, KLEINER DE. Liver histology: Diagnostic and prognostic features[J]. Clin Liver Dis, 2020, 24(1): 61-74. DOI: 10.1016/j.cld.2019.09.004.
[2]SHENNAR HK, AL-ASMAR D, KADDOURA A, et al. Diagnosis and clinical features of organic acidemias: A hospital-based study in a single center in Damascus, Syria[J]. Qatar Med J, 2015, 2015(1): 9. DOI: 10.5339/qmj.2015.9.
[3]VOCKLEY J, ANDERSSON HC, ANTSHEL KM, et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline[J]. Genet Med, 2014, 16(2): 188-200. DOI: 10.1038/gim.2013.157.
[4]BOVE KE. 7-histologic patterns of metabolic liver diseases[M]//SAXENA R. Practical hepatic pathology: a diagnostic approach (Second Edition). Philadelphia; Elsevier. 2018: 101-124.
[5]KIMURA A, KAGAWA T, TAKEI H, et al. Rotor syndrome: Glucuronidated bile acidemia from defective reuptake by hepatocytes[J]. Hepatol Commun, 2021, 5(4): 629-633. DOI: 10.1002/hep4.1660.
[6]HADZIYANNIS S, GERBER MA, VISSOULIS C, et al. Cytoplasmic hepatitis B antigen in “ground-glass” hepatocytes of carriers[J]. Arch Pathol, 1973, 96(5): 327-330.
[7]DESDENTADO L, ESPERT R, SANZ P, et al. Lafora disease: a review of the literature[J]. Rev Neurol, 2019, 68(2): 66-74.
[8]OSHEA AM, WILSON GJ, LING SC, et al. Lafora-like ground-glass inclusions in hepatocytes of pediatric patients: a report of two cases[J]. Pediatr Dev Pathol, 2007, 10(5): 351-357. DOI: 10.2350/06-12-01948.1.
[9]SUMMAR ML, MEW NA. Inborn errors of metabolism with hyperammonemia: Urea cycle defects and related disorders[J]. Pediatr Clin North Am, 2018, 65(2): 231-246. DOI: 10.1016/j.pcl.2017.11.004.
[10]MATSUMOTO S, HBERLE J, KIDO J, et al. Urea cycle disorders-update[J]. J Hum Genet, 2019, 64(9): 833-847. DOI: 10.1038/s10038-019-0614-4.
[11]
BIGOT A, TCHAN MC, THOREAU B, et al Liver involvement in urea cycle disorders: a review of the literature[J]. J Inherit Metab Dis, 2017, 40(6): 757-769. DOI: 10.1007/s10545-017-0088-5.
[12]HICKS J, WARTCHOW E, MIERAU G. Glycogen storage diseases: a brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment[J]. Ultrastruct Pathol, 2011, 35(5): 183-196. DOI: 10.3109/01913123.2011.601404.
[13]CHAPMAN J, ARNOLD JK. Reye syndrome[M]. StatPearls. Treasure Island (FL); StatPearls Publishing, Copyright 2022, StatPearls Publishing LLC. 2022.
[14]
CORPECHOT C, BARBU V, CHAZOUILLRES O, et al. Genetic contribution of ABCC2 to Dubin-Johnson syndrome and inherited cholestatic disorders[J]. Liver Int, 2020, 40(1): 163-174. DOI: 10.1111/liv.14260.
[15]MORAIS M, COUVERT P, JRU I, et al. Rotor syndrome presenting as Dubin-Johnson syndrome[J]. Case Rep Gastroenterol, 2022, 16(2): 452-455. DOI: 10.1159/000525517.
[16]
MARUO Y, NAKAHARA S, YANAGI T, et al. Genotype of UGT1A1 and phenotype correlation between Crigler-Najjar syndrome type II and Gilbert syndrome[J]. J Gastroenterol Hepatol, 2016, 31(2): 403-408. DOI: 10.1111/jgh.13071.
[17]
BHANDARI J, THADA PK, YADAV D. Crigler Najjar syndrome[M]. StatPearls. Treasure Island (FL); StatPearls Publishing, Copyright 2022, StatPearls Publishing LLC. 2022.
[18]Vanishing bile duct syndrome[M]. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD); National Institute of Diabetes and Digestive and Kidney Diseases. 2012.
[19]
TYAGI I, PURI AS, SAKHUJA P, et al. Co-occurrence of cytomegalovirus-induced vanishing bile duct syndrome with papillary stenosis in HIV infection[J]. Hepatol Res, 2013, 43(3): 311-314. DOI: 10.1111/j.1872-034X.2012.01058.x.
[20]
SHIVARAM PS, GIRISH KP. Alagille syndrome and the liver: current insights[J]. Euroasian J Hepatogastroenterol, 2018, 8(2): 140-147. DOI: 10.5005/jp-journals-10018-1280.
[21]ROMI L, SAXEN A. Practical hepatic pathology: A diagnostic approach[M]. Elsevier, 2011.
[22]
ANGELICO R, SENSI B, MANZIA TM, et al. Chronic rejection after liver transplantation: Opening the Pandoras box[J]. World J Gastroenterol, 2021, 27(45): 7771-7783. DOI: 10.3748/wjg.v27.i45.7771.
[23]
KMEID M, LIU X, BALLENTINE S, et al. Idiopathic non-cirrhotic portal hypertension and porto-sinusoidal vascular disease: Review of current data[J]. Gastroenterology Res, 2021, 14(2): 49-65. DOI: 10.14740/gr1376.
[24]XANTHOPOULOS A, STARLING RC, KITAI T, et al. Heart failure and liver disease: Cardiohepatic interactions[J]. JACC Heart Fail, 2019, 7(2): 87-97. DOI: 10.1016/j.jchf.2018.10.007.
[25]
SHUKLA A, SHRESHTHA A, MUKUND A, et al. Budd-Chiari syndrome: consensus guidance of the Asian Pacific Association for the study of the liver (APASL)[J]. Hepatol Int, 2021, 15(3): 531-567. DOI: 10.1007/s12072-021-10189-4.
[26]
LUO WP, MA H, ZHAO XY. Pathological features of hepatic vascular diseases and key points in differential diagnosis[J]. J Clin Hepatol, 2018, 34(11): 2289-2294. DOI: 10.3969/j.issn.1001-5256.2018.11.005.
羅文萍, 马红, 赵新颜. 肝血管病的病理学特征及鉴别诊断要点[J]. 临床肝胆病杂志, 2018, 34(11): 2289-2294. DOI: 10.3969/j.issn.1001-5256.2018.11.005.
收稿日期:
2023-01-11;录用日期:2023-02-18
本文编辑:林姣
猜你喜欢
上海农业学报(2017年4期)2017-04-10 12:40:34
新教育时代·教师版(2016年30期)2016-12-05 12:54:21
中国中药杂志(2016年20期)2016-11-19 12:33:47
今日健康(2016年12期)2016-11-17 19:20:27
今日健康(2016年12期)2016-11-17 14:47:33
中国科技博览(2016年19期)2016-10-19 13:05:18
中国科技博览(2016年18期)2016-10-19 10:10:52
科技视界(2016年21期)2016-10-17 18:28:05
中国实用医药(2016年24期)2016-10-17 06:23:23
医学研究杂志(2015年6期)2015-07-01 17:40:14
