
磷酸弗林酸性簇分选蛋白2对血管紧张素Ⅱ诱导的心肌肥大的影响及其机制
2023-04-29 00:44杨福情敖翔肖丹丹刘丙岩王建勋宋林
精准医学杂志 2023年5期
杨福情 敖翔 肖丹丹 刘丙岩 王建勋 宋林

[摘要] 目的 探究磷酸弗林酸性簇分选蛋白2(PACS-2)对血管紧张素Ⅱ(AngⅡ)诱导的心肌肥大的影响及其机制。方法 剪取并消化出生1~2 d的SD大鼠乳鼠的心脏组织,采用差速贴壁法获取乳鼠心肌细胞(NRCMs),原代培养24 h。以浓度为1.5 μmol/L的AngⅡ处理NRCMs 0、3、6、12、24 h,采用Western blot方法检测细胞中FUN14域蛋白1(FUNDC1)、PACS-2、三磷酸肌醇受体蛋白(IP3R)的表达水平,采用实时荧光定量PCR(RT-qPCR)方法检测细胞中心钠肽(ANP)、脑钠肽(BNP)、β-肌球蛋白重链(β-MHC)mRNA的表达水平。将NRCMs分为A~C组,A组使用无血清培养基培养,B、C组分别转染si-NC和si-PACS-2,采用Western blot方法检测各组细胞中PACS-2蛋白的表达水平。将NRCMs分为D~G组,D组使用无血清培养基培养,E组以浓度0.15 μmol/L的AngⅡ培养,F、G组分别转染si-NC、si-PACS-2后再以浓度0.15 μmol/L的AngⅡ培养,采用TRITC-鬼笔环肽染色技术检测各组心肌细胞表面积,RT-qPCR检测各组细胞ANP、BNP、β-MHC mRNA的表达水平,以Fluo-4,AM探针检测各组细胞胞质Ca2+的水平。将NRCMs分为H~K组,H组使用无血清培养基培养,I、J组分别转染si-NC、si-PACS-2,K组转染si-PACS-2并且以浓度1 μmol/L的钙调蛋白(CaM)拮抗剂处理后,采用RT-qPCR方法检测各组细胞ANP、BNP、β-MHC mRNA的表达水平。结果 以浓度1.5 μmol/L的AngⅡ处理NRCMs 0、3、6、12、24 h,NRCMs中ANP、BNP、β-MHC mRNA的表达水平呈时间依赖性上调(F=25.73~58.30,P<0.05),并且处理第24小时时相较于第0小时时均显著上调(t=5.35~37.50,P<0.05)。以浓度1.5 μmol/L的AngⅡ处理NRCMs 0、3、6、12、24 h,NRCMs中IP3R、PACS-2以及FUNDC1蛋白的表达水平均呈时间依赖性下调(F=5.37~9.07,P<0.05),并且处理第24小时时相较于第0小时时显著下调(t=6.55~7.42,P<0.05)。与B组相比,C组NRCMs中PACS-2蛋白的表达水平显著下调(t=5.92,P<0.05)。与F组相比,G组NRCMs中心肌细胞表面积增大,ANP、BNP、β-MHC mRNA的表達水平及胞质Ca2+水平均上调(t=3.50~26.60,P<0.05)。与J组相比,K组NRCMs中ANP、BNP、β-MHC mRNA表达水平均显著下调(t=3.27~5.13,P<0.05)。结论 敲低PACS-2可增加NRCMs中胞质Ca2+水平,并且可能以Ca2+-CaM依赖的方式加重AngⅡ诱导的心肌肥大的发生。
[关键词] 磷酸弗林酸性簇分选蛋白2;心脏扩大;内质网;线粒体膜;钙;血管紧张素Ⅱ
[中图分类号] R541
[文献标志码] A
EFFECT OF PHOSPHOFURIN ACIDIC CLUSTER SORTING PROTEIN 2 ON ANGIOTENSIN Ⅱ-INDUCED CARDIAC HYPERTROPHY AND ITS MECHANISM \ YANG Fuqing, AO Xiang, XIAO Dandan, LIU Bingyan, WANG Jianxun, SONG Lin(Department of Biochemistry and Molecular Biology Basic Medicine, Qingdao University, Qingdao 266021, China)
[ABSTRACT] Objective To investigate the effect of phosphofurin acidic cluster sorting protein 2 (PACS-2) on angiotensin Ⅱ (Ang Ⅱ)-induced cardiac hypertrophy and its mechanism. Methods The heart tissue of neonatal Sprague-Dawley rats aged 1-2 d was clipped and digested, and the differential adhesion method was used to obtain neonatal rat cardiomyocytes (NRCMs), which were primary cultured for 24 h. After NRCMs were treated with 1.5 μmol/L Ang Ⅱ for 0, 3, 6, 12, and 24 h, Western blot was used to measure the protein expression levels of FUN14 domain-containing protein 1 (FUNDC1), PACS-2, and inositol 1,4,5-trisphosphate receptor (IP3R) in cells, and RT-qPCR was used to measure the mRNA expression levels of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC) in cells. NRCMs were divided into groups A-C, group A was cultured with serum-free medium, and groups B and C were transfected with si-NC and si-PACS-2, respectively, Western blot was used to measure the protein expression level of PACS-2 in each group. NRCMs were divided into groups D-G, group D was cultured with serum-free medium, group E was cultured with 0.15 μmol/L Ang Ⅱ, and groups F and G were transfected with si-NC and si-PACS-2, respectively, and were then cultured with 0.15 μmol/L Ang Ⅱ, TRITC-phalloidin staining was used to measure the surface area of cardiomyocytes in each group, RT-qPCR was used to measure the mRNA expression levels of ANP, BNP, and β-MHC in each group, and Fluo-4,AM probe was used to measure the level of cytosolic Ca2+in each group. NRCMs were divided into groups H-K, group H was cultured with serum-free medium, groups I and J were transfected with si-NC and si-PACS-2, respectively, and group K was transfected with si-PACS-2 and cultured by 1 μmol/L CaM inhibitor; RT-qPCR was used to measure the mRNA expression levels of ANP, BNP, and β-MHC in each group. Results After treatment of NRCMs with Ang Ⅱ at a concentration of 1.5 μmol/L for 0, 3, 6, 12, and 24 h, the mRNA expression levels of ANP, BNP, and β-MHC in NRCMs gradually increased over time (F=25.73-58.30,P<0.05), and the levels at 24 hours of treatment were significantly upregulated compared with those at 0 hour (t=5.35-37.50,P<0.05). After treatment of NRCMs with Ang Ⅱ at a concentration of 1.5 μmol/L for 0, 3, 6, 12, and 24 h, the protein expression levels of IP3R, PACS-2, and FUNDC1 in NRCMs gradually decreased over time (F=5.37-9.07,P<0.05), and the levels at 24 h of treatment were significantly downregulated compared with those at 0 h (t=6.55-7.42,P<0.05). Compared with group B, group C had a significant reduction in the protein expression level of PACS-2 in NRCMs (t=5.92,P<0.05). Compared with group F, group G had significant increases in the surface area of cardiomyocytes, the mRNA expression levels of ANP, BNP, and β-MHC, and the level of cytosolic Ca2+in NRCMs (t=3.50-26.60,P<0.05). Compared with group J, group K had significant reductions in the mRNA expression levels of ANP, BNP, and β-MHC in NRCMs (t=3.27-5.13,P<0.05). Conclusion Knockdown of PACS-2 can increase the level of cytosolic Ca2+in NRCMs and aggravate Ang Ⅱ-induced cardiac hypertrophy in a Ca2+-CaM-dependent manner.
[KEY WORDS] Phosphofurin acidic cluster sorting protein 2; Cardiomegaly; Endoplasmic reticulum; Mitochondrial membranes; Calcium; Angiotensin Ⅱ
病理性心肌肥大是由各种损伤刺激因子和持续压力负荷引起的代偿性心脏扩张[1]。长期病理性心肌肥大是心肌梗死、心律失常、心力衰竭甚至猝死的重要原因[2]。病理性心肌肥大的发病机制之一是心肌细胞Ca2+动态紊乱,而胞质中Ca2+超载能激活Ca2+依赖相关肥大信号通路[3-4]。线粒体相关内质网膜(MAMs)是内质网(ER)和线粒体之间的动态膜结构[5],与细胞内钙稳态及线粒体稳态等密切相关[6-7]。大量研究表明MAMs异常与多种心血管疾病有关[8]。磷酸弗林酸性簇分选蛋白2(PACS-2)作为MAMs上的结构蛋白,通过影响线粒体分裂调节MAMs的形成[9]。然而,PACS-2在心肌肥大中的作用机制尚不是很清楚。本研究旨在探讨敲低乳鼠心肌细胞(NRCMs)中的PACS-2对AngⅡ诱导的心肌肥大的影响,为预防和治疗心肌肥大甚至心力衰竭提供理论依据。
1 材料和方法
1.1 实验动物和材料
SD大鼠乳鼠购自青岛大任富城畜牧公司;胞质钙离子探针(Fluo-4,AM)购自上海翌圣生物科技有限公司;血管紧张素Ⅱ(AngⅡ)购自上海阿拉丁生化科技股份有限公司;TRITC标记鬼笔环肽染料购自北京索莱宝科技有限公司;FUN14域蛋白1(FUNDC1)、PACS-2、三磷酸肌醇受体蛋白(IP3R)兔单克隆抗体购自英国Abcam公司;钙调蛋白(CaM)拮抗剂购自美国MCE公司;PACS-2小干扰RNA购自上海吉玛制药技术有限公司。
1.2 实验方法
1.2.1 细胞提取和培养 将30只出生1~2 d的SD大鼠乳鼠置于无菌玻璃皿当中,使用体积分数0.75的乙醇清洗2次,无菌条件下解剖分离乳鼠心脏,PBS缓冲液(pH 7.4)清洗;切碎并置于1.2 g/L胰液素和0.14 g/LⅡ型胶原酶配制的消化液中;消化后的細胞悬液在37 ℃下孵育1 h,上清液即为NRCMs。将稀释至1×109个/L的NRCMs接种于含有DMEM-F12培养基的孔板(含体积分数0.05 FBS,10 g/L的青链霉素混合液)中,置于37 ℃、含体积分数0.05 CO2恒温培养箱中培养24 h,用于后续实验。
1.2.2 实时荧光定量PCR(RT-qPCR)方法检测细胞中心钠肽(ANP)、脑钠肽(BNP)、β-肌球蛋白重链(β-MHC)mRNA的表达水平 将经过分离的NRCMs接种于6孔板内培养24 h,待细胞汇合度约达70%时,再经无血清培养基饥饿培养12 h后,于培养基中加入浓度1.5 μmol/L的AngⅡ,分别培养0、3、6、12、24 h后,采用Trizol法提取细胞的总RNA,NanoDrop分光光度计测定RNA的浓度与纯度。按照反转录试剂盒说明书步骤将1 μg RNA反转录为cDNA,继而以此为模板进行RT-qPCR。采用2-△△CT方法来计算目的基因的相对表达量。ANP、BNP、β-MHC 以GAPDH为内参照,引物名称及其序列见表1。
1.2.3 Western blot方法检测细胞中IP3R、PACS-2、FUNDC1蛋白表达水平 按1.2.2中的方法将NRCMs培养0、3、6、12、24 h后,每孔中加入RIPA裂解液,冰上静置10 min,提取细胞总蛋白。蛋白样品以含150 g/L的分离胶以及50 g/L浓缩胶的SDS-PAGE凝胶电泳分离后,转膜1.5 h,并使用含体积分数0.05的脱脂牛奶封闭1 h。封闭结束后,一抗4 ℃孵育过夜,洗膜后二抗室温孵育1 h,使用超敏ECL发光液显影。使用Image J软件分析相关蛋白的灰度值,以β-Actin为内参照,目的蛋白的相对表达水平以目的蛋白灰度值/内参照蛋白灰度值计算,作为NRCMs中IP3R、PACS-2、FUNDC1蛋白的相对表达水平。
1.2.4 Western blot方法检测A~C组转染后的NRCMs细胞中PACS-2蛋白的表达水平 将分离的NRCMs接种于6孔板培养24 h,汇合度约70%时,经无血清培养基饥饿培养12 h后,分为A~C组。A组使用无血清培养基培养24 h,B、C组分别用LipofectamineTM3000转染试剂将si-NC(B组)和si-PACS-2(C组)转染NRCMs,6 h后换液,继续培养18 h后提取细胞总蛋白,采用Western blot方法检测A~C组NRCMs细胞中PACS-2蛋白的表达水平。
1.2.5 TRITC-鬼笔环肽染色检测D~G组细胞骨架大小 将分离的NRCMs接种于24孔板内培养24 h,汇合度约达50%时,经无血清培养基饥饿培养12 h后,分为D~G组。D组使用无血清培养基培养48 h;E组培养基中加入浓度0.15 μmol/L的AngⅡ培养24 h;F、G组分别转染NRCMs si-NC、si-PACS-2并培养24 h后,于培养基中均加入浓度0.15 μmol/L的AngⅡ培养24 h。培养结束后各组去除原培养基,PBS洗涤3次后,按TRITC-鬼笔环肽染色试剂盒说明书要求,加入TRITC-鬼笔环肽(100 nmol/L)工作液,37 ℃下避光孵育30 min,并用双光子共聚焦显微镜(德国Leica公司)观察细胞骨架。用Image J软件测量F-肌动蛋白染色细胞的表面积。
1.2.6 RT-qPCR方法检测D~G组细胞中ANP、BNP、β-MHC mRNA的表达水平 取6孔板中培养48 h的D~G组NRCMs,RT-qPCR方法检测D~G组细胞中ANP、BNP、β-MHC mRNA的表达水平,3种基因均以GAPDH作为内参照。
1.2.7 Fluo-4,AM检测D~G组细胞胞质中Ca2+的水平 取6孔板当中培养48 h以后的D~G组NRCMs,按照Fluo-4钙离子检测试剂盒说明书要求,去除原培养基,PBS洗涤3次后,加入Fluo-4,AM(2 μmol/L)工作液,37 ℃下避光孵育30 min,并通过倒置荧光显微镜(德国Leica公司)观察。用Image J软件分析细胞内钙离子的荧光强度,以此代表细胞胞质中Ca2+的水平。
1.2.8 RT-qPCR方法检测H~K组细胞中ANP、BNP以及β-MHC mRNA的表达水平 将分离后的NRCMs接种于6孔板内培养24 h,汇合度约达70%时,经无血清培养基饥饿培养12 h以后,分为H~K组。H组使用无血清培养基培养48 h;I、J组分别转染NRCMs si-NC、si-PACS-2并且培养24 h;K组培养基中加入浓度1 μmol/L的CaM拮抗剂,培养24 h后转染si-PACS-2后再培养24 h。培养结束以后,采用RT-qPCR方法检测H~K组细胞中的ANP、BNP、β-MHC mRNA表达水平,3种基因均是以GAPDH为内参照。
1.3 统计学处理
使用Graph Pad Prism 8.01软件进行统计分析。所有实验均重复3次,结果取均值。计量资料以x?±s表示,两组间比较采用t检验;多组比较采用单因素方差分析,两两比较采用LSD-t检验。以P<0.05为差异有统计学意义。
2 结果
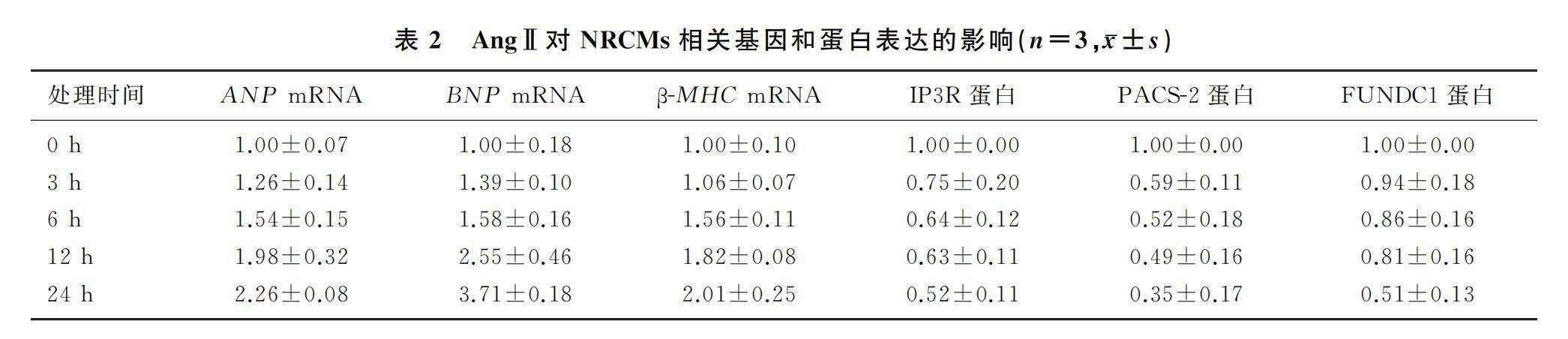
2.1 AngⅡ对NRCMs相关基因和蛋白表达影响
以浓度为1.5 μmol/L的AngⅡ处理NRCMs 0、3、6、12及24 h以后,NRCMs中ANP、BNP、β-MHC mRNA的表达水平比较差异具有显著统计学意义(F=25.73~58.30,P<0.05)。其中在加入1.5 μmol/L的AngⅡ处理NRCMs第6、12、24小时时相较于处理第0小时时,上述3种肥大基因表达水平差异有显著性(t=4.46~37.50,P<0.05)。见表2。选择以上肥大基因在AngⅡ处理NRCMs第24小时时差异最显著(P值最小)时间点进行后续实验。
以浓度为1.5 μmol/L的AngⅡ处理NRCMs 0、3、6、12、24 h后,NRCMs中IP3R、PACS-2以及FUNDC1蛋白的表达水平比较差异均具有显著性(F=5.37~9.07,P<0.05);其中加入1.5 μmol/L的AngⅡ处理NRCMs第24小时时相较于处理第0小时时,上述3个指标进行比较,差异均具有显著性(t=6.55~7.42,P<0.05)。见表2。
2.2 A~C组NRCMs中PACS-2蛋白相对表达量比较
Western blot检测结果显示,A~C组NRCMs当中PACS-2蛋白的表达水平分别为1.00±0.00、0.94±0.06和0.57±0.12,各组比较差异有显著性(F=24.40,P<0.05);与B组相比,C组中PACS-2蛋白的表达水平显著降低(t=5.92,P<0.05)。
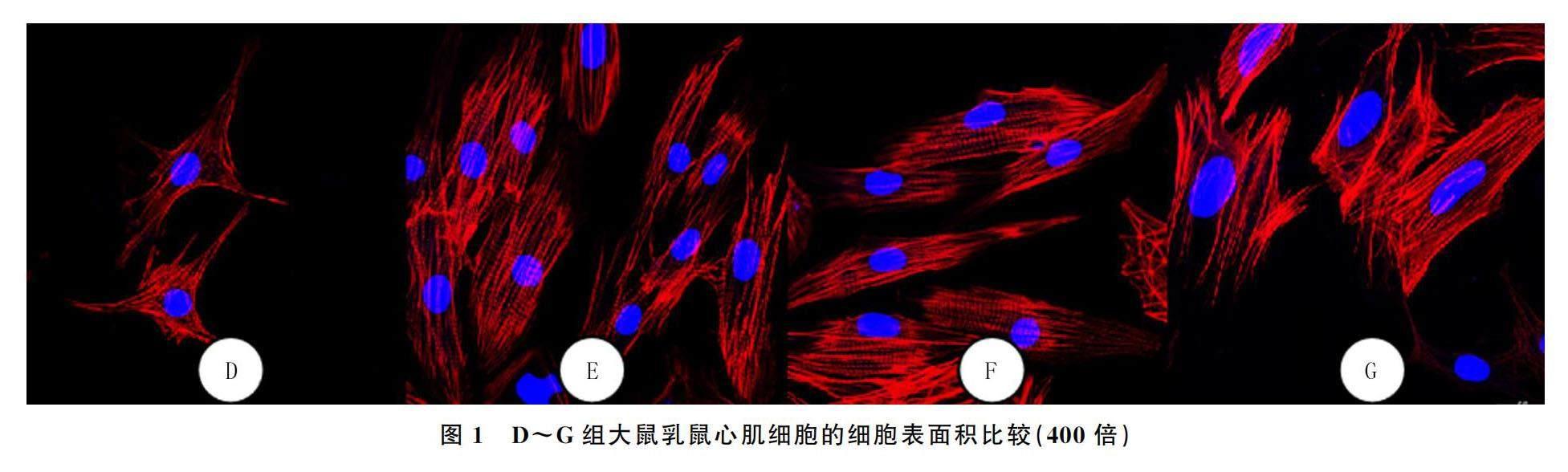
2.3 D~G组NRCMs的细胞表面积以及ANP、BNP、β-MHC mRNA的表達水平比较
经AngⅡ和转染si-PACS-2处理后,D~G组NRCMs的细胞表面积比较差异均具有显著性(F=679.4,P<0.05),其中与E、F组进行比较,D、G组NRCMs的细胞表面积差异具有显著性(t=11.92~26.52,P<0.05)。见图1。
D~G组NRCMs中ANP、BNP以及β-MHC mRNA的表达水平比较差异均具有显著意义(F=12.55~42.96,P<0.05)。与D组相比较,E、F组NRCMs中上述3种肥大基因的表达水平差异无显著性(P>0.05);与E组相比,F组NRCMs中上述3种肥大基因的表达水平差异无显著性(P>0.05);与E、F组相比,G组NRCMs中上述3种肥大基因的表达水平差异具有显著性(t=2.74~15.18,P<0.05)。见表3。
2.4 D~G组NRCMs中胞质Ca2+水平比较
Fluo-4,AM检测结果显示,D~G组NRCMs中Ca2+相对表达水平分别为1.00±0.07、1.91±0.24、1.98±0.23和2.61±0.25,各组NRCMs中胞质Ca2+水平比较差异显著(F=29.51,P<0.05)。与E、F组相比,D、G组NRCMs中胞质Ca2+水平差异具有显著性(t=5.35~8.25,P<0.05);与E组相比,F组NRCMs中胞质Ca2+水平差异无显著性(P>0.05)。
2.5 H~K组NRCMs中的ANP、BNP以及β-MHC mRNA表达水平比较
经过转染si-PACS-2以及以CaM蛋白抑制剂(1 μmol/L)处理NRCMs后,H~K组NRCMs中ANP、BNP、β-MHC mRNA的表达水平比较差异均有显著性(F=14.69~18.69,P<0.05);与J组相比,H、I、K组NRCMs中上述3种肥大基因的表达水平差异具有显著性(t=3.27~8.31,P<0.05);与H组进行比较,I组NRCMs中上述3种肥大基因的表达水平比较差异均无显著意义(P>0.05)。见表4。
3 讨论
心肌肥大是心肌由于各种因素引起的机械负荷增加而产生的代偿性反应,其可逐渐发展为失代偿性,导致不可逆转的心力衰竭甚至猝死[10-11]。因此
表4 H~K组心肌肥大基因的相对表达水平的比较(n=3,x?±s)分组ANP mRNABNP mRNAβ-MHC mRNAH组1.00±0.301.00±0.091.00±0.14I组0.96±0.150.91±0.141.20±0.23J组2.44±0.391.92±0.212.27±0.44K组1.16±0.381.30±0.250.91±0.11
预防和逆转心肌肥大对于心血管疾病的治疗具有重要临床意义。
MAMs作为内质网和线粒体的关键接触位点,对细胞生理功能的调节发挥着重要作用。研究发现MAMs异常与缺血再灌注、心力衰竭等多种心血管疾病有关[12-13]。MAMs富集多种连接和功能蛋白,如FUNDC1、PACS-2及IP3R等[14-15]。FUNDC1與IP3R蛋白结合形成连接ER和线粒体的桥梁,参与MAMs的形成[16]。此外,PACS-2作为一种新的分选蛋白,参与ER和线粒体之间的结合[7]。本研究将NRCMs经浓度1.5 μmol/L的AngⅡ处理0、3、6、12、24 h,RT-qPCR和Western blot方法检测结果显示,心肌肥大基因ANP、BNP、β-MHC的表达水平均呈时间依赖性上调,其中各肥大基因在AngⅡ处理NRCMs第24小时时相比处理第0小时时差异最显著。同时,IP3R、FUNDC1、PACS-2蛋白的表达水平均呈现了时间依赖性下调,其中PACS-2蛋白的表达水平在AngⅡ处理24 h后下调最为明显。以上的结果均表明,AngⅡ处理能抑制NRCMs中MAM相关蛋白的表达,诱导心肌肥大。真核细胞内存在多种蛋白降解的途径,包括泛素化降解、细胞自噬以及非编码RNA靶向导致的降解等,探究PACS-2蛋白在心肌肥大中下降的机制及其具体的调控机制,对于心肌肥大发病机制及其新的治疗方案的研究至关重要。PACS-2作为MAMs的关键结构蛋白之一,其缺失会破坏MAMs结构和功能完整性[17-18]。本研究中以高浓度(1.5 μmol/L)的AngⅡ处理NRCMs以后,心肌肥大基因ANP、BNP、β-MHC的表达水平均呈时间依赖性上调,并且细胞质中PACS-2蛋白呈现时间依赖性下调,提示PACS-2蛋白是心肌肥大的负调节剂。为了进一步探究PACS-2的功能,本研究通过转染si-PACS-2以敲低NRCMs中PACS-2基因的表达,并采用低浓度(0.15 μmol/L)AngⅡ处理NRCMs,以说明敲低PACS-2对NRCMs发生心肌肥大的敏感性的影响。NRCMs经转染si-PACS-2 24 h后,给予低浓度0.15 μmol/L 的AngⅡ处理24 h以后,分别进行TRITC-鬼笔环肽染色以及RT-qPCR检测,结果显示,低浓度的AngⅡ就可以促进NRCMs细胞表面积增大,但是敲低PACS-2能够促进AngⅡ诱导的NRCMs细胞表面积明显增大;同时,低浓度AngⅡ对NRCMs中ANP、BNP、β-MHC基因表达无显著影响,而敲低PACS-2能促进AngⅡ诱导这3种心肌肥大基因的表达明显上调。以上结果说明,敲低PACS-2可提高NRCMs对AngⅡ诱导的心肌肥大的敏感性。
病理性心肌肥大的发病机制极其复杂,在病理性肥大的心脏中常常出现线粒体功能障碍、纤维化增加以及Ca2+稳态失调[1,19]。研究发现,内质网和线粒体之间的Ca2+转移需要PACS-2进行介导,而PACS-2的下降能导致内质网向胞浆释放Ca2+增加[7,18]。因此,敲低PACS-2提高NRCMs对AngⅡ诱导的心肌肥大的敏感性的具体机制可能涉及细胞中的Ca2+稳态失调。本研究针对NRCMs转染si-PACS-2再培养24 h后,给予低浓度AngⅡ处理24 h,行Fluo-4,AM染色,结果显示,低浓度AngⅡ能促进NRCMs胞质Ca2+水平显著增加,而敲低PACS-2使得这种现象更为明显。该结果说明了敲低PACS-2提高了NRCMs对于AngⅡ诱导的胞质Ca2+增加的敏感性。在心肌肥大早期,PACS-2的下降能诱导半胱氨酸蛋白酶8水解B细胞受体相关蛋白31产生p20亚基,而后者引起Ca2+从内质网释放到线粒体中[20]。本研究通过敲低NRCMs中PACS-2的表达,检测到的NRCMs胞质Ca2+水平显著增加,这种现象可能发生于心肌肥大晚期。因此,检测心肌肥大不同时期时细胞内Ca2+水平,可以揭示在心肌肥大整个的发病过程中,PACS-2对细胞内Ca2+调节的具体机制。研究表明PACS-2参与内质网与线粒体之间Ca2+的运输,可能与其介导的载货蛋白的膜运输有关[21]。因此,后续应该进一步研究在内质网与线粒体之间PACS-2介导的Ca2+转运的具体过程,从而了解PACS-2在心肌肥大中的调控机制。
胞质Ca2+是参与心肌肥大的重要信号分子,主要通过两种信号转导通路机制诱导核内肥大基因转录,包括钙/钙调蛋白依赖性蛋白激酶Ⅱ-组蛋白去乙酰化酶通路、钙调神经磷酸酶-活化T细胞核因子(NFAT)通路等[22-23]。有研究报道,G蛋白偶联受体激酶5能以Ca2+-CaM依赖的方式激活NFAT,促进肥大基因的转录[24-25]。因此推测敲低PACS-2也以同样的方式参与了心肌肥大的发生。本研究针对NRCMs给予CaM拮抗剂处理24 h后,AngⅡ(0.15 μmol/L)处理24 h,RT-qPCR检测结果显示,敲低PACS-2能促进ANP、BNP、β-MHC心肌肥大基因的上调,而CaM拮抗剂抑制了这种现象。以上结果说明,敲低PACS-2可以Ca2+-CaM依赖的方式介导心肌肥大的发生。
綜上所述,敲低PACS-2能增加NRCMs中胞质Ca2+水平,并且是以Ca2+-CaM依赖的方式加重AngⅡ诱导的心肌肥大的发生。但是PACS-2如何通过调节胞质Ca2+发挥心脏保护作用还需要进一步探究。本研究为心肌肥大及持续性心肌肥大导致的心血管疾病预防和治疗提供了有效实验数据。
作者声明:杨福情、敖翔、肖丹丹、刘丙岩参与了研究设计;杨福情、王建勋、宋林参与了论文的写作和修改。所有作者均阅读并同意发表该论文,且声明不存在利益冲突。
[参考文献]
[1]NAKAMURA M, SADOSHIMA J. Mechanisms of physiolo-gical and pathological cardiac hypertrophy[J]. Nat Rev Car-diol, 2018,15(7):387-407.
[2]AUBDOOL A A, THAKORE P, ARGUNHAN F, et al. A novel α-calcitonin gene-related peptide analogue protects against end-organ damage in experimental hypertension, car-diac hypertrophy, and heart failure[J]. Circulation, 2017,136(4):367-383.
[3]ZOU Y Z, LIANG Y Y, GONG H, et al. Ryanodine receptor type 2 is required for the development of pressure overloa-dinduced cardiac hypertrophy[J]. Hypertension, 2011,58(6):1099-1110.
[4]ZHAO G J, ZHAO C L, OUYANG S, et al. Ca2+-dependent NOX5 (NADPH oxidase 5) exaggerates cardiac hypertrophy through reactive oxygen species production[J]. Hypertension, 2020,76(3):827-838.
[5]CSORDS G, RENKEN C, VRNAI P, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria[J]. J Cell Biol, 2006,174(7):915-921.
[6]PERRONE M, CAROCCIA N, GENOVESE I, et al. The role of mitochondria-associated membranes in cellular homeostasis and diseases[J]. Int Rev Cell Mol Biol, 2020,350:119-196.
[7]YANG M. Mitochondria-associated membranes (MAMs):A novel therapeutic target for treating metabolic syndrome[J]. Curr Med Chem, 2021,28(7):1347-1362.
[8]GAO P, YAN Z C, ZHU Z M. Mitochondria-associated endoplasmic reticulum membranes in cardiovascular diseases[J]. Front Cell Dev Biol, 2020,8:604240.
[9]SIMMEN T, ASLAN J E, BLAGOVESHCHENSKAYA A D, et al. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis[J]. EMBO J, 2005,24(4):717-729.
[10]OKA T, AKAZAWA H, NAITO A T, et al. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure[J]. Circ Res, 2014,114(3):565-571.
[11]GUO J, MIHIC A, WU J, et al. Canopy 2 attenuates the transition from compensatory hypertrophy to dilated heart fai-lure in hypertrophic cardiomyopathy[J]. Eur Heart J, 2015,36(37):2530-2540.
[12]PAILLARD M, TUBBS E, THIEBAUT P A, et al. Depres-sing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury[J]. Circulation, 2013,128(14):1555-1565.
[13]WALTERS A M, PORTER G A Jr, BROOKES P S. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy[J]. Circ Res, 2012,111(9):1222-1236.
[14]HAYASHI T, RIZZUTO R, HAJNOCZKY G, et al. MAM: More than just a housekeeper[J]. Trends Cell Biol, 2009,19(2):81-88.
[15]HERNNDEZ-ALVAREZ M I, SEBASTIN D, VIVES S, et al. Deficient endoplasmic reticulum-mitochondrial phosphatidylserine transfer causes liver disease[J]. Cell, 2019,177(4):881-895.
[16]WU S N, LU Q L, WANG Q L, et al. Binding of FUN14 domain containing 1 with inositol 1, 4, 5-trisphosphate receptor in mitochondria-associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo[J]. Circulation, 2017,136(23):2248-2266.
[17]ARRUDA A P, PERS B M, PARLAKG?L G, et al. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity[J]. Nat Med, 2014,20(12):1427-1435.
[18]YU S J, ZHANG L P, LIU C, et al. PACS2is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca2+elevation[J]. Exp Cell Res, 2019,379(2):191-202.
[19]VAN BERLO J H, MAILLET M, MOLKENTIN J D. Signaling effectors underlying pathologic growth and remodeling of the heart[J]. J Clin Invest, 2013,123(1):37-45.
[20]BRECKENRIDGE D G, STOJANOVIC M, MARCELLUS R C, et al. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol[J]. J Cell Biol, 2003,160(7):1115-1127.
[21]LI C R, LI L, YANG M, et al. PACS-2:A key regulator of mitochondria-associated membranes (MAMs)[J]. Pharmacol Res, 2020,160:105080.
[22]KUMAR S, WANG G, LIU W J, et al. Hypoxia-induced mitogenic factor promotes cardiac hypertrophy via calcium-dependent and hypoxia-inducible factor-1α mechanisms[J]. Hypertension, 2018,72(2):331-342.
[23]LV L F, LI T Y, LI X L, et al. The lncRNA Plscr4 controls cardiac hypertrophy by regulating miR-214[J]. Mol Ther Nucleic Acids, 2018,10:387-397.
[24]HULLMANN J E, GRISANTI L A, MAKAREWICH C A, et al. GRK5-mediated exacerbation of pathological cardiac hypertrophy involves facilitation of nuclear NFAT activity[J]. Circ Res, 2014,115(12):976-985.
[25]COLEMAN R C, EGUCHI A, LIEU M, et al. A peptide of the N terminus of GRK5 attenuates pressure-overload hypertrophy and heart failure[J]. Sci Signal, 2021,14(676):eabb5968.
(本文編辑 耿波 厉建强)
猜你喜欢
中国临床医学影像杂志(2019年1期)2019-04-25
电子制作(2019年24期)2019-02-23
西南交通大学学报(2018年5期)2018-11-08
苏州科技大学学报(自然科学版)(2017年1期)2017-03-20
知识产权(2016年8期)2016-12-01
天津医药(2016年9期)2016-10-20
中西医结合心脑血管病杂志(2016年20期)2016-03-01
山东医药(2015年14期)2016-01-12
中国老年学杂志(2015年16期)2015-03-05
癌变·畸变·突变(2015年4期)2015-02-27
