
颜色转换(C^N)2Ir(pic-X)配合物结构及光谱性质的量子理论
2023-04-29 18:25聂建航曲珈慧曾妮白福全张建坡金丽
吉林大学学报(理学版) 2023年2期
聂建航 曲珈慧 曾妮 白福全 张建坡 金丽
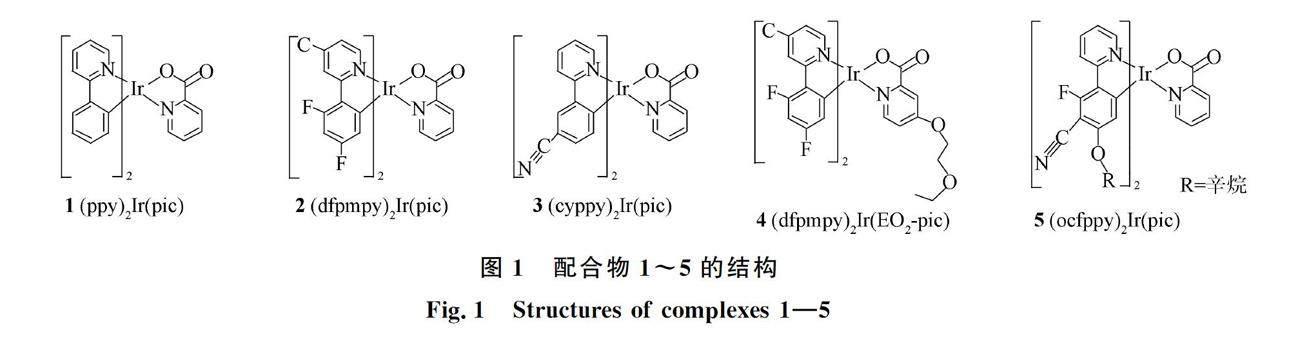


摘要: 采用B3LYP和UB3LYP方法分別优化一系列(C^N)2Ir(pic-X)(C^N=ppy(1), dfpmpy(2), cyppy(3), dfpmpy(4), ocfppy(5), ppy=苯基吡啶,dfpmpy=2-(2,4-双氟苯基)-4-甲基吡啶,cyppy=2-(3-氰基苯基)吡啶,ocfppy=2-(2-辛氧基-3-氰基-4-氟苯基)吡啶, pic=2-羧基吡啶,X=H(1,2,3,5)和EO2(4),EO2=4-二乙氧基)配合物的S0和T1态几何结构. 利用含时密度泛函理论(TD-DFT)方法, 结合Gauss程序中的溶剂化模型得到它们在CHCl3溶剂中的光谱特征. 结果表明: 所有结构参数和光谱数据均接近它们的实验值; 配合物1~5最低能的吸收和发射分别出现在459,415,412,397,393 nm和567,532,544,491,490 nm处; 其最高占据分子轨道(HOMOs)主要占据在Ir原子和C^N配体上,配合物1,2,3,5的最低空轨道(LUMOs)由pic配体贡献,配合物4由C^N和pic两个配体共同占据,因此它们具有不同的金属到配体及配体到配体的电荷转移跃迁(MLCT/LLCT)特征. 可见通过调取代基团π电子捐赠能力可改变配合物的发光颜色.
关键词: 铱(Ⅲ)配合物; 颜色转换; 发光材料; 光谱特征
中图分类号: O643 文献标志码: A 文章编号: 1671-5489(2023)02-0419-07
Quantum Theory of Structures and Spectral Properties of Color Conversion (C^N)2Ir(pic-X) Complexes
NIE Jianhang1, QU Jiahui1, ZENG Ni1, BAI Fuquan2, ZHANG Jianpo1, JIN Li1(1. School of Chemical and Pharmaceutical Engineering, Jilin Institute of Chemical Technology, Jilin 132022,Jilin Province, China; 2. College of Chemistry, Jilin University,Changchun 130021,China)
收稿日期: 2022-05-05.
第一作者简介: 聂建航(1998—),男,汉族,硕士研究生,从事过渡金属发光材料量子理论的研究, E-mail: njh2023@126.com.
通信作者简介: 张建坡(1980—),男,汉族,博士,教授,从事发光材料理论设计的研究, E-mail: zhangjp725@126.com; 金 丽(1980—),女,汉族,博士,教授,从事光功能材料合成及理论的研究,E-mail: canoe8013@126.com.
基金项目: 国家自然科学基金青年科学基金(批准号: 21405058).
Abstract: The geometries of S0 and T1 states of a series of iridium(Ⅲ) complexes (C^N)2Ir(pic-X) (C^N=ppy(1), dfpmpy(2), cyppy(3), dfpmpy(4), ocfppy(5), ppy=Phenylpyridine, dfpmpy=2-(2,4-difluorophenyl)-4-methylpyri-dine, cyppy=2-(3-cyanophenyl)- pyridine, ocfppy=2-(2-octyl-3-cyano-4-fluorophenyl)pyridine; pic=2-carboxyl-pyridine; X=H(1,2,3,5), EO2(4), EO2=4-diethyloxy) were optimized by the B3LYP and UB3LYP methods, respectively. Time dependent density functional theory (TD-DFT) method together with the solvation model in Gauss program were used to obtain their spectral properties in CHCl3 solvent. The results show that the structural parameters and spectral datas are close to their experimental values. The lowest energy absorptions and phosphorescence emissions are at 459,415,412,397,393 nm,and 567,532,544,491,490 nm, respectively. The highest occupied molecular orbital (HOMOs) of complexes 1—5 are mainly localized on the Ir atom and C^N ligands, the lowest unoccupied molecular orbital (LUMOs) are mainly contributed by the pic ligand for complexes 1,2,3,5, and dominantly localized on the C^N and pic ligands for complex 4. Therefore, they have different transition characteristics of metal to ligand and ligand to ligand charge transfer (MLCT/LLCT). The calculation results show that the phosphorescence color can be changed by altering the π electron-donating ability of substituent group.
Keywords: iridium(Ⅲ) complex; color conversion; luminescent material; spectral property
近年來,由于过渡金属配合物[1]在化学传感[2]、 全彩显示[3]以及生物分子探针[4]等领域的广泛应用,因此已引起人们广泛关注. 在众多配合物中,铱(Ir)配合物具有量子效率高、 激发态寿命短和可覆盖整个可见光谱的可调发射波长等优点[5-7]. 其中由红、 绿、 蓝三基色合成的白色有机发光二极管(WOLED)在全彩显示器和固态灯具中应用广泛[8-9]. 已合成的绿光铱配合物(ppy)2Ir(tmd)(ppy=苯基吡啶, tmd=四甲基庚烷酮酸盐)的量子效率达32.3%[10],红光铱配合物[(nbt)2Ir(pic-Cz)](nbt=2-(1-萘基)苯并噻唑, pic=吡啶甲酸, Cz=9-(4-(溴甲基)苯基)-9H-咔唑)的量子效率大于30%[11],但蓝光铱配合物通常表现出较低的效率和较短的寿命,成为制约光电显示行业发展的短板.
目前,(dfpmpy)2Ir(pic)(dfpmpy=2-(2,4-双氟苯基)-4-甲基吡啶)是被广泛研究的蓝光配合物之一[12-13],但其存在两个缺陷: 1) 在普通溶液中的溶解度较差; 2) 基于其理论基础上的设备寿命较短. 为克服这两个缺陷,研究人员对该类配合物进行了合成,Kozhevnikov等[14]合成了(ocfppy)2Ir(pic) (ocfppy=2-(2-辛氧基,3-氰基,4-氟苯基)吡啶)配合物,并探究其物理化学性质. 研究表明,引入ocfppy辅助配体可显著提高配合物的溶解度,且在293 K,脱气1,2-二氯乙烷中测得的激发态寿命可达到微秒级. 为进一步揭示该类配合物的发光规律,本文采用量子化学方法,对5个含有苯基吡啶和吡啶甲酸配体铱配合物(C^N)2Ir(pic-X)(C^N=ppy(1), dfpmpy(2), cyppy(3), dfpmpy(4), ocfppy(5), cyppy=2-(3-氰基苯基)吡啶,X=H(1,2,3,5)和EO2(4),EO2=4-二乙氧基)进行理论研究.
1 计算方法
图1为5个(C^N)2Ir(pic-X)配合物的分子结构,在整个优化过程中所有分子均无对称性限制. 采用密度泛函方法中的B3LYP[15] 和UB3LYP[16]泛函,分别优化配合物1~5的S0和T1几何结构. 在S0和T1态结构的基础上,利用含时密度泛函(TDDFT)[17]方法,结合选定的溶剂模型,获得了其在CHCl3溶剂中的光谱特征. 计算中,对Ir原子采用17个价电子的准相对论赝势模型,其他原子采用6-31G(d)基函, 该计算方法常用于计算过渡金属配合物发光材料,方法准确、 可靠[18-19]. 所有计算均在曙光A620-G30服务器上由Gauss09(D.01)[20]程序包完成.
2 结果与讨论
2.1 S0和T1态结构
图2为配合物3的优化结构及主体结构原子编号,以配合物3为例,其他配合物的主体部分原子编号与其相对应.
表1~表3分别为配合物1~5的优化S0和T1态的部分键长、 键角和二面角参数. 由表1可见,配合物5的基态Ir—C(1)和Ir—O(1)键长分别为0.202 2,0.217 7 nm,与其实验值分别相差0.003 6,0.005 2 nm. 由表2可见,键角C(1)—Ir—N(2)的理论与实验值相差0.5°,表明计算结果符合真实实验分子. 由5个配合物的键长、 键角和二面角可见,引入取代基对配合物的主体结构影响较小,取代基团不能决定分子主体的电子云排布. 由表2和表3可见,键角N(2)—Ir—N(3)接近180°,表明3个原子基本在一条直线上,二面角C(1)—Ir—N(2)—C(2)和C(1)—Ir—N(2)—O(1)均接近90°,即3个双齿配体基本垂直,表明配合物具有紧凑的正八面体结构,符合六配位金属配合物的结构形式. 配合物5的键角和二面角与其他4个配合物相比差距较大,表明引入的辛氧基团产生了空间位阻效应,导致配合物的结构发生一定程度的扭曲.
5个配合物在激发态的Ir—N(1)键长分别延长了约0.001 6~0.008 4 nm,Ir—C(1)和Ir—O(1)键长缩短了(0.001~0.003 nm),所有键角和二面角均发生较小的变化(<2.4°),表明激发态下电子跃迁使电子云由金属偏向ppy配体和pic配体的羧基一端,从而增强它们与金属的相互作用, 消弱了金属与pic配体吡啶环的相互作用. 受到空间位阻的影响,配合物5的部分键角和二面角也呈现出与其他配合物不同的变化趋势.
2.2 边界分子轨道成分及能量
表4列出了5个配合物的重要边界分子轨道成分和能量. 由表4可见,5个配合物的最高占据分子轨道(HOMOs)主要占据在金属Ir和C^N配体上,配合物1,2,3,5的最低未占空轨道(LUMOs)为占据在pic-X配体上的π*型轨道,只有配合物4的LUMO仍有较高比例的C^N配体成分,表明其最低能吸收与其他配合物具有不同的跃迁本质. 比较它们的前线占据分子轨道可见,C^N配体在所有轨道上均有较高比例的占据,而在非占据分子轨道上C^N和pic-X配体互有贡献,因此可认为C^N配体为该类配合物的主配体,在跃迁过程中起主要作用. 若单独考虑5个配合物取代基团(—CH3,—CN,—F,—OR)的轨道占据情况,只有配合物5在部分边界分子轨道上有较高比例(<30%),表明较大的取代基团除了空间位阻效应外,还可部分影响配合物的跃迁性质.
由图3可见,与配合物1相比,随着引入吸电子基团,配合物2,3,5的HOMO和LUMO能量同時降低,但由于HOMO降低更多,因此HOMO-LUMO能级间隙变大,导致对应最低能吸收和发射波长蓝移. 在配合物4中加入了乙氧基团,导致其在降低HOMO能量的同时提升了LUMO能量,这也使得HOMO-LUMO能级间隙变大,但能级差变大的原因与配合物2,3,5的原因不同.
2.3 吸收光谱
根据S0态几何结构,利用含时密度泛函方法得到配合物1~5在CHCl3溶剂中的吸收光谱,为直观地表述配合物的紫外可见光谱特征,图4为理论模拟的Gauss型吸收曲线,
挑选各配合物典型吸收数据列于表5.
由图4和表5可见,配合物1~5均有3个明显的吸收区间,其最低能吸收分别位于459(1),415(2),412(3),397(4),393(5) nm处,
均来自HOMO→LUMO的激发,振子强度非常低. 由表5可见,配合物1的HOMO主要由49.7%d(Ir)和40.4% π(ppy)配体成分构成,LUMO由94.2%的pic配体占据,因此该跃迁被指认为[d(Ir)+π(ppy)→π*(pic)]的金属Ir到pic配体和ppy配体到pic配体的电荷转移(MLCT/LLCT)跃迁. 配合物2,3,5与1的该跃迁具有相似的轨道占据和跃迁性质. 配合物4在397 nm处的跃迁与以上配合物略有不同,其LUMO有56.2%的dfpmpy配体成分,该跃迁虽具有MLCT/LLCT特征,但其MLCT含有较高比例的金属Ir到dfpmpy配体跃迁,这与其他4个配合物的跃迁不同. 配合物4和5的该跃迁实验值分别为377,374 nm,与理论值相差约20 nm,这是由于溶剂化模型的选择或多或少偏离分子的真实情况所致,在误差允许范围内.
由各配合物的最低能吸收可见,在ppy配体的苯环上增加—F和—CN等吸电子基团,可使其最低能吸收发生一定程度的蓝移,且随吸电子能力越强,蓝移的趋势越大,如配合物1和5波长差达到66 nm,这足以改变配合物的发光颜色. 在不同位置添加给电子基团,除影响配合物的吸收波长,还对跃迁性质产生一定的影响,且不同的取代位置影响效果不同. 如配合物4与2相比波长蓝移18 nm,并使跃迁性质发生部分改变,配合物4与5相比波长变化较小,表明pic配体上苯环的4-取代位置较敏感,针对该位置的合成将有较大的应用潜力.
配合物1~5的第二个代表性吸收分别位于351(1),339(2),333(3),331(4),332(5) nm处,均来自HOMO-2→LUMO激发,其振子强度远高于最低能吸收,在实验上易于观测. 配合物1~4的跃迁初始轨道和终止轨道占据情况与最低能吸收基本相同,因此该处跃迁也具有MLCT/LLCT跃迁特征. 由于配合物5的HOMO-2有高达80.7%的ocfppy配体成分,因此该跃迁被指认为[π(ocfppy)+d(Ir)→π*(pic)]的电荷转移跃迁,具有主要的LLCT跃迁特征,并伴有少量的MLCT跃迁微扰. 表明辛氧基团的参与改变了配合物5的跃迁性质.
配合物的高能吸收分别在262(1),232(2),236(3),238(4),233(5) nm处,这些位置的振子强度最大. 它们的跃迁性质主要分为两类,其中: 配合物1,3,4的该跃迁只发生在C^N配体上,具有典型的C^N配体内部的电荷转移(ILCT)跃迁特征; 配合物2和5的跃迁初始轨道具有较高比例的pic配体成分(>55%), 因此该跃迁以LLCT跃迁为主,并存在少量的ILCT跃迁. 比较5个配合物的第二和第三个典型吸收,除配合物1外,其他4个配合物波长非常接近(差值<8 nm),表明吸电子或给电子基团对这两个吸收带的影响较小.
2.4 磷光发射光谱
表6为配合物1~5在CHCl3溶液中T1态的磷光发射光谱. 由表6可见,5个配合物的磷光发射分别位于567(1),532(2),544(3),491(4),490(5) nm处,均来自LUMO→HOMO跃迁,并与最低能吸收具有相似的轨道占据和跃迁性质. 且配合物4仍具有区别于其他配合物的3MLCT/3LLCT特征. 配合物4和5的理论与实验值之差小于5 nm,表明理论计算较准确. 比较配合物1~5的最低能吸收和发射的能量,其差分别为0.51(1),0.65(2),0.73(3),0.60(4),0.62(5) eV ,这是由它们基态到激发态结构改变, 导致Stokes频移所致. 上述研究表明,通过改变配体上的吸电子或给电子取代基团可实现该类配合物的颜色转换.
综上所述,本文研究了5个不同取代基团铱[(C^N)2Ir(pic-X)]配合物的几何结构和光谱特征,它们的键长、 键角和二面角均较接近,表明加入取代基团并未影响配合物的主体结构. 当加入较大的取代基团时,可产生明显的空间位阻效应,配合物5的几何结构发生明显扭曲. 在苯基吡啶配体的苯环上加入吸电子基团,可同时降低HOMO和LUMO能量,导致HOMO-LUMO能级间隙变大,最低能吸收和发射波长蓝移. 在羧基吡啶配体的吡啶环4号位置加入给电子基团,在降低HOMO能量的同时提升LUMO能量,也能增加HOMO-LUMO能级间隙,表明该类配合物的吡啶环4号位置较敏感,有利于开展配合物的分子设计. 此外,加入取代基团可不同程度影响配合物的轨道占据和跃迁性质,在配合物1和2的3个吸收带上均有体现,但不能从根本上改变配合物的跃迁性质. 本文研究表明,通过配合物的分子设计可实现绿光到蓝光材料的有效开发.
参考文献
[1] 支晓彤,姜伟丽,李继聪,等. 双金属催化剂用于烯烃氢甲酰化反应的研究进展 [J]. 天然气化工—C1化学与化工,2022,47(1): 15-23. (ZHI X T, JIANG W L, LI J C, et al. Research Progress of Bimetallic Catalysts for Hydroformylation of Olefifins [J]. Natural Gas Chemical Industry, 2022,47(1): 15-23.)
[2] NIE J, LI J P, DENG H, et al. Progress on Click Chemistry and Its Application in Chemical Sensors [J]. Chinese Journal of Analytical Chemistry, 2015, 43(4): 609-617.
[3] JUSTINO C I L, FREITAS A C, PEREIRA R, et al. Recent Developments in Recognition Elements for Chemical Sensors and Biosensors [J]. TrAC Trends in Analytical Chemistry, 2015, 68: 2-17.
[4] ALGHOUL E, BASBOUS J, CONSTANTINOU A. An Optogenetic Proximity Labeling Approach to Probe the Composition of Inducible Biomolecular
Condensates in Cultured Cells [J]. STAR Protocols, 2021, 2(3): 100677-1-100677-16.
[5] RSUSCH A F, THOMPSON M E, YERSIN H. Matrix Effects on the Triplet State of the OLED Emitter Ir(4,6-dFppy)2(pic)(FIrpic): Investigations by
High-Resolution Optical Spectroscopy [J]. Inorganic Chemistry, 2009, 48(5): 1928-1937.
[6] NAZEERUDDIN M K, HUMPHRY-BAKER R, BERNER D, et al. Highly Phosphorescence Iridium Complexes and Their Application in Organic Light-Emitting Devices [J]. Journal of the American Chemical Society, 2003, 125(29): 8790-8797.
[7] 宋鵬飞,侯建国,王秀林. 可再生能源氢储能与氢转化利用技术及发展模式分析 [J]. 天然气化工—C1化学与化工,2022,47(3): 26-32. (SONG P F, HOU J G, WANG X L. Analysis of Hydrogen Energy Storage for Renewables and Hydrogen Conversion Technology and Development model [J]. Natural Gas Chemical Industry, 2022,47(3): 26-32.)
[8] KAMTEKAR K T, MONKMAN A P, BRYCE M R. Recent Advances in White Organic Light-Emitting Materials and Devices (WOLEDs) [J]. Advanced
Materials, 2010, 22(5): 572-582.
[9] ZHOU G J, WANG Q, WANG X Z, et al. Metallophosphors of Platinum with Distinct Main-Group Elements: A Versatile Approach towards Color Tuning and
White-Light Emission with Superior Efficiency/Color Quality/Brightness Trade-Offs [J]. Journal of Materials Chemistry, 2010, 20(35): 7472-7484.
[10] KIM K H, MOON C K, LEE J H, et al. Highly Efficient Organic Light-Emitting Diodes with Phosphorescent Emitters Having High Quantum Yield and
Horizontal Orientation of Transition Dipole moments [J]. Advanced Materials, 2014, 26(23): 3844-3847.
[11] YU T Z, BAO Y J, ZHAO Y L, et al. Synthesis, Photo- and Electro-Luminescence of Red-Emitting Ir(Ⅲ) Complexes with 2-(1-Naphthyl)benzothiazole and Carrier
Transporting Group-Functionalized Picolinate Ligands [J]. Journal of Organometallic Chemistry, 2016, 825/826: 33-40.
[12] LAMANSKY S, DJUROVICH P, MURPHY D, et al. Highly Phosphorescent Bis-cyclometalated Iridium Complexes: Synthesis, Photophysical [J]. Journal
of the American Chemical Society, 2001, 123(18): 4304-4312.
[13] CHO W, SARADA G, PARK J S, et al. Synthesis and Characterization of Blue-Emitting Ir(Ⅲ) Complexes with Multi-functional Ancillary Ligands for
Solution-Processed Phosphorescent Organic Light-Emitting Diodes [J]. Organic Electronics, 2014, 15(10): 2328-2336.
[14] KOZHEVNIKOV V N, DAHMS K, BRYCE M R. Nucleophilic Substitution of Fluorine Atoms in 2,6-Difluoro-3-(pyridin-2-yl) Benzonitrile Leading to
Soluble Blue-Emitting Cyclometalated Ir(Ⅲ) Complexes [J]. Journal of Organic Chemistry, 2011, 76(12): 5143-5148.
[15] BECKE A D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior [J]. Physical Review A,1988,38(6): 3098-3100.
[16] FOREMAN J B,HEAD G M, POPLE A. Toward a Systematic Molecular Orbital Theory for Excited States [J]. Journal of Physical Chemistry, 1992, 96:
135-149.
[17] BARONE V, COSSI M. A New Definition of Cavities for the Computation of Solvation Free Energies by the Polarizable Continuum Model [J]. Journal of
Chemical Physics, 1997, 107: 3210-3221.
[18] ZHANG J P, WANG Y, MA J B, et al. Density Functional Theory Investigation on Iridium(Ⅲ) Complexes for Efficient Blue Electrophosphorescence [J].
RSC Advances, 2018, 8(35): 19437-19448.
[19] 張建坡,周欣,白福全,等. 一类[Os(Ⅱ)(CO)3(tfa)(L)](L=O^O,O^N,N^N)配合物的结构和光谱特征 [J]. 物理化学学报,2008,24(12): 2243-2248. (ZHANG J P, ZHOU X, BAI F Q, et al. Structures and Spectroscopic Properties for a Series of [Os(Ⅱ)(CO)3(tfa)(L)] (L=O^O,O^N,N^N) Complexes [J]. Acta Physico-Chimica Sinica, 2008, 24(12): 2243-2248.)
[20] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 03, Revision C.02 [CP]. Wallingford, CT: Gaussian Inc, 2004.
(责任编辑: 单 凝)
