
基于线粒体COI和D-loop序列的黑龙江流域蛇种群遗传结构分析
2023-03-29 04:22胡宗云杨培民宋红梅牟希东
淡水渔业 2023年2期
胡宗云,杨培民,宋红梅,牟希东
(1.辽宁省淡水水产科学研究院,辽宁省水生动物病害防治重点实验室,辽宁辽阳 111000;2.中国水产科学研究院珠江水产研究所,农业农村部休闲渔业重点实验室,广州 510380)
线粒体DNA分子标记是核基因组分子标记的重要补充,已广泛用于水产动物的系统发育、生物地理学和保护生物学研究[15]。细胞色素氧化还原酶I(COI) 基因位于mtDNA的蛋白质编码区,进化速率适中,在物种分子系统演化和分类中应用较多;而D-loop序列属于非编码区,是mtDNA进化速度最快的区域,常用于鱼类进化生物学和群体遗传学研究。本研究测定、分析了黑龙江流域3个蛇群体的COI和D-loop序列,研究了其遗传多样性水平和群体间遗传分化程度,以期为东北地区蛇种质资源保护提供理论依据。
1 材料与方法
1.1 材料
2020年4月至2021年5月,采用地笼网和挂网采集了3个群体88尾野生蛇样本,其中抚远群体30尾(黑龙江水系,标记为FY)、饶河群体29尾(乌苏里江水系,标记为RH)、榆树群体29尾(松花江水系,标记为YS)(表1)。样本体长为13~23 cm,体重为16~51 g。每尾剪取鳍条1~2 g,保存于95%酒精内带回实验室。
表1 蛇样品来源信息
Tab.1 The source information on samples of longnose gudgeon S.dabryi Bleeker

表1 蛇样品来源信息
群体所属河流取样时间采集地经纬度样品数FY黑龙江2020年4月黑龙江省佳木斯市抚远市浓桥镇 N48°11'58.36″;E134°17'1.28”30RH乌苏里江2020年5月黑龙江省双鸭山市饶河县八五九农场 N47°20'13.50″; E134°9'10.42″29YS松花江2021年5月吉林省长春市榆树市培英街道N44°50'13.35″ ;E126°37'30.50″29
1.2 方法
1.2.1 DNA提取、PCR扩增及测序
取鳍条0.1g左右,利用天根生化科技有限公司的基因组提取试剂盒(DP304),按照试剂盒说明书提取总DNA。COI序列扩增引物为C-F(5′-TC AACCAACCACAAAGACATTGGCAC-3′)和C-R(5′-TAGACTTCTGGGTGGCCAAAGAATCA-3′)[16],D-loop序列扩增引物为D-F(5′-CTAACTCCCAAAGC TAGAATTCT-3′)和D-R(5′-ATCTTAGCATCTTCAGTG-3′)[17],上述引物均由生工生物(上海)有限公司合成。PCR反应体系为25 μL:含2.5 μL 10×PCR buffer(Mg2+plus,20 mmol/L)、2 μL dNTP mix(2.5 mmol/L)、0.25 μL Taq DNA polymerase(5 U/μL)、上下游引物各1 μL(10 μmol/L)、模板1 μL,双蒸水补足至25 μL。PCR扩增条件为:94 ℃预变性5 min;35个循环,每个循环包括94 ℃变性1 min,56 ℃退火1 min,72 ℃延伸1 min 30 s;最后73 ℃总延伸10 min。本研究所有的PCR反应均在ABI 9700扩增仪上进行,扩增产物经1.2%琼脂糖凝胶电泳检测,获得目的条带的产物于-20 ℃冰箱内保存备用。PCR产物送至生工生物(上海)有限公司进行纯化,并由ABI 3730全自动测序仪双向测序,测序引物为上述PCR扩增引物。
1.2.2 数据处理
测得的序列经BioEdit编辑去除两端低质量的碱基,然后在NCBI网站上进行比对,留取与蛇线粒体基因组(KF534790.1、KF612272.1)COI和D-loop序列同源性99%以上的序列。运用MEGA 6.0软件包中的Alignment by ClustalW程序对留取的序列进行比对和剪切,并计算序列碱基组成、变异位点数和转换与颠换值;采用邻接法(Neighbor-Joining),基于Kimura双参数法(Kimura 2-parameter,K2p)模型构建单倍型进化树。利用DnaSP 6.0软件统计两个基因的单倍型数、单倍型多样性(h)、平均核苷酸差异数(K)及核苷酸多样性(π)。应用Arlequin 3.5 软件进行中性检验,并用其分子方差分析(AMOVA)方法计算遗传变异在群体内和群体间的分布及群体间遗传分化系数(Fst),1 000次重复随机抽样重排后进行显著性检验,以P<0.05作为差异显著性水平。利用Popart 1.7软件构建TCS单倍型网络图。
2 结果
2.1 序列组成与遗传多样性分析
COI和D-loop序列经比对剪齐后,分别获得了长度为582~583 bpCOI和834~835 bp D-loop的同源序列(表2)。COI同源序列中共检测出15个变异位点,其中简约信息位点12个,单突变位点3个,一共定义了13个单倍型;平均碱基组成:A=28.7%、T=23.2%、G=28.9%、C=19.2%,A+T含量(52%)大于G+C含量(48%);平均转换/颠换值为9.8。D-loop同源序列中共检测到14个变异位点,包括9个简约信息位点和5个单突变位点,定义单倍型15个,平均碱基组成:A=32%、T=31.9%、G=14.4%、C=21.8%,A+T含量(64%)大于G+C含量(36%);平均转换/颠换值为4.6。COI和D-loop碱基组成均呈较明显的AT偏好。
3个群体COI基因片段总体单倍型多样性、核苷酸多样性和核苷酸差异数分别为0.377 3、0.000 9和0.510 4;各群体的单倍型多样性指数为0.330 7~0.406 2,单倍型多样性指数大小顺序为YS>RH>FY;核苷酸多样性指数均为0.000 9(表2)。3个群体D-loop序列总体单倍型多样性、核苷酸多样性和核苷酸差异数分别为0.577 0、0.000 9和0.780 4;各群体的单倍型多样性指数和核苷酸多样性指数分别介于0.492 0~0.692 0和0.000 8~0.001 1;群体间D-loop单倍型多样性指数、核苷酸多样性指数大小顺序一致,均为YS>RH>FY。3个群体基于COI和D-loop序列的遗传多样性指数相近,单倍型多样性、核苷酸多样性指数处于较低水平,样本间两个序列的核苷酸差异较小。
表2 蛇3个群体COI 和D-loop基因的遗传多样性参数
Tab.2 Genetic diversity parameters of COI and D-loop genes in three populations of longnose gudgeon S.dabryi Bleeker

表2 蛇3个群体COI 和D-loop基因的遗传多样性参数
遗传多样性参数COIFYRHYS总体D-loopFYRHYS总体序列长度582^583582^583582^583582^583835835834^835834^835变异位点数5771568914单一突变位点数46733565简约突变位点数110123339单倍型数67513791015单倍型多样性0.330 70.404 30.406 20.377 30.492 00.545 00.692 00.577 0核苷酸多样性0.000 90.000 90.000 90.000 90.000 80.001 00.001 10.000 9平均核苷酸差异数k0.500 00.511 80.520 00.510 40.627 00.831 00.872 00.780 4
2.2 遗传距离和遗传分化
用Kimura双参数法(Kimura 2-parameter,K2p)计算3个群体间遗传距离和遗传分化指数如表3所示。基于COI序列的分析结果显示,3个群体的群体内和群体间遗传距离相等,均为0.000 9;3个群体的群体间遗传分化指数-0.004 9~0.007 6。基于D-loop序列的计算结果表明,3个群体的群体内遗传距离为0.008 0~0.010 0,大小顺序为RH =YS >FY;群体间遗传距离为0.000 9~0.001 0,YS与FY群体间遗传距离和FY与RH群体间遗传距离相等(0.000 9),略小于YS与RH群体间的遗传距离(0.001 0);3个群体间的遗传分化指数介于-0.002 8~0.011 4。3个群体基于COI和D-loop序列的遗传距离、遗传分化指数较小,预示3个群体间遗传差异较小,遗传分化不明显。
表3 蛇遗传分化指数(对角线上)、群体间遗传距离(对角线下)和群体内遗传距离(对角线)
Tab.3 Pairwise Fst(above diagonal),genetic distance between(below diagonal) and within(diagonal) populations of longnose gudgeon S.dabryi Bleeker

表3 蛇遗传分化指数(对角线上)、群体间遗传距离(对角线下)和群体内遗传距离(对角线)
群体FYRHYSFY0.000 9-0.004 9-0.003 4COIRH0.000 90.000 90.007 6YS0.000 90.000 90.000 9FY0.000 80.011 4-0.002 8 D-loopRH0.000 90.001 00.006 8YS0.000 90.001 00.001 0
基于COI序列的分子方差分析(AMOVA)显示,群体内遗传变异占遗传变异的100.02%,远大于群体间遗传变异(-0.02%);基于D-loop序列AMOVA分析同样显示,群体内的遗传变异占比(99.49%)也远大于群体间遗传变异(0.51%)。基于COI和D-loop序列的分子方差分析表明,3个蛇群体整体遗传变异几乎全部来自群体内,群体间遗传变异占比极小(表4)。
表4 蛇3个群体COI和D-loop基因的AMOVA分析
Tab.4 The AMOVA analysis of COI and D-loop genes in three populations of longnose gudgeon S.dabryi Bleeker
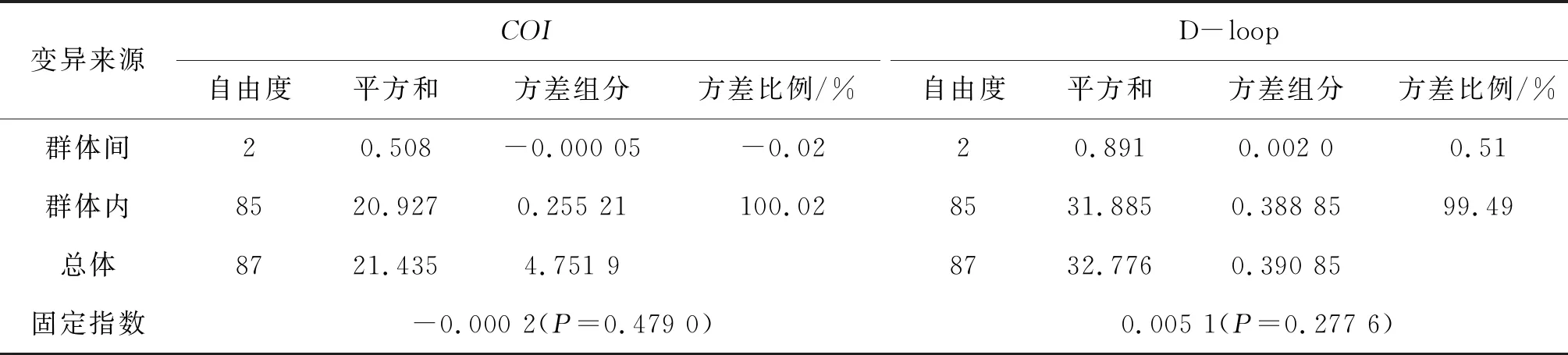
表4 蛇3个群体COI和D-loop基因的AMOVA分析
变异来源COI自由度平方和方差组分方差比例/%D-loop自由度平方和方差组分方差比例/%群体间20.508-0.000 05-0.0220.8910.002 00.51群体内8520.9270.255 21100.028531.8850.388 8599.49总体8721.4354.751 98732.7760.390 85固定指数-0.000 2(P=0.479 0)0.005 1(P=0.277 6)
2.3 单倍型聚类分析与分布
88个COI序列共检测到13个单倍型(图1),Hap1和Hap3为3个群体共享单倍型,在所测样本中出现频率分别为79.55%和4.55%,其中Hap1在每个群体占比均最高,属于优势单倍型;Hap9为FY和YS的共享单倍型,其余单倍型为3个群体特有的单倍型,FY、RH和YS群体特有单倍型的数量分别为3、5和2。88个D-Loop序列共定义了15个单倍型(图2),Hap2、Hap7和Hap9为3个群体共享单倍型,其中Hap2数量达到58个,占样本总数的65.91%,属于优势单倍型;Hap11、Hap12为FY与YS的共享单倍型,Hpa1、Hap8为RH与FY的共享单倍型,Hap6为FY和RH的共享单倍型,其余单倍型为各群体特有单倍型,特有单倍型数量1-3个不等。

图1 蛇3个群体COI基因单倍型的NJ树及在各群体中的分布
2.4 种群历史动态


图4 蛇COI(A)和D-loop序列的核苷酸错配分布图
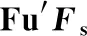
表5 基于COI和D-loop序列的Tajima′D和检验
3 讨论
3.1 蛇群体遗传多样性
单倍型多样性(h)和核苷酸多样性(π)是评价种群遗传多样性的重要指标,也是衡量物种或种群线粒体DNA变异程度的重要指标。本研究3个蛇群体基于D-Loop序列的单倍型多样性和核苷酸多样性分别为0.492 0~0.692 0、0.000 8~0.001 1,低于嘉陵江蛇种群(h:0.875~0.940;π:0.010 0~0.264 0)[4],也低于近缘种斑点蛇(S.punctatus)(h:0.703~0.960;π:0.021 7~0.048 2)[4]和长鳍吻(Rhinogobioventralis))(h:0.640~0.837;π:0.001 0~0.0.01 5)[18],这些差异除与物种的遗传特性有关外,还与样本量大小、序列长短、分析序列的位置有关。本研究88个蛇样本基于COI序列的遗传多样性指数(h =0.377 3;π=0.000 9)略小于D-loop(h=0.577 0;π=0.000 9),这可能与COI和D-loop的进化速率不同有关:COI和D-loop分别位于线粒体的编码区和非编码区,COI受到的选择压力大于D-loop,进化速度相对较慢,发生变异的概率相对低一些,相似的结果在银鲳(Pampusargenteus)[19]、强弹涂鱼(Periophthalmuscantonensis)[20]、鲤(Cyprinuscarpio)[21]等鱼类上亦有报道。根据GRANT等[22]设定h=0.5,π=0.005阈值,本研究3个蛇群体基于COI和D-loop序列的遗传多样性指数类型分别属于低h低π型、高h低π型,考虑到核苷酸多样性在一定程度上比单倍型多样性更能反映群体的遗传多样性[22], 3个蛇群体遗传多样性处于较低水平,应加强该区域蛇的种质资源保护。
3.2 蛇群体遗传分化
群体间遗传距离、遗传分化指数与群体分化程度为线性关系,是衡量群体分化程度的重要指标[23]。3个蛇群体基于两个序列的群体间遗传距离与群体内的遗传距离均较小,大小范围相互重叠且处于一个水平,群体间遗传距离远低于0.05种群这一标准[24],这说明3个群体间遗传差异较小。群体间的遗传分化指数小于0.05,表明群体间遗传分化极小[25]。AMOVA分析表明,群体内的变异远大于群体间变异,3个蛇群体总的遗传变异主要来自群体内,预示3个蛇群体亲缘关系较近。此外,本研究基于COI和D-loop序列的单倍型NJ系统发育树和网络图显示,3个蛇群体共享单倍型较多,特有单倍型交错地聚集在一起或分布在祖先单倍型周围,没有形成明显的地理谱系,也印证了群体间遗传分化较小的观点。上述结果可能与蛇栖息环境和繁殖习性有关:本研究中 3个群体的采样点分布于黑龙江水系的支流,河流间处于连通状态,采样点之间未形成严格意义上的地理隔离;而蛇产漂流性卵,鱼卵或鱼苗随水流进行扩散[1],利于群体扩散。换言之,敞开性水域和较强的扩散能力促进了3个蛇群体的基因交流,使群体间遗传分化较小。相似的结果在蛇长江种群[26]、长鳍吻(R.ventralis)[18]、圆筒吻(Rhinogobiocylindricus)[27]等亚科鱼类上亦有报道。鉴于3个群体遗传距离较小、遗传分化水平较低这一现状,渔业资源管理实践中可以将其设为一个管理单元(MU)。
3.3 蛇群体历史动态
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
海南医学(2020年1期)2020-01-18
电子制作(2019年24期)2019-02-23
广西林业科学(2016年3期)2016-03-16
中国康复理论与实践(2015年10期)2015-12-24
百科知识(2015年18期)2015-09-10
