
5-mRNA-based prognostic signature of survival in lung adenocarcinoma
2023-03-17 01:39QianLinXiaXiaoMengHeYanMaQiuYueLiYuZhenDuJinWang
Qian-Lin Xia,Xiao-Meng He,Yan Ma,Qiu-Yue Li,Yu-Zhen Du,Jin Wang
Qian-Lin Xia,Yu-Zhen Du,Laboratory Medicine,Shanghai Sixth People's Hospital Affiliated to Shanghai Jiao Tong University School of Medicine,Shanghai 200233,China
Xiao-Meng He,Yan Ma,Qiu-Yue Li,Jin Wang,Scientific Research Center,Shanghai Public Health Clinical Center,Fudan University,Shanghai 201508,China
Abstract BACKGROUND Lung adenocarcinoma(LUAD)is the most common non-small-cell lung cancer,with a high incidence and a poor prognosis.AIM To construct effective predictive models to evaluate the prognosis of LUAD patients.METHODS In this study,we thoroughly mined LUAD genomic data from the Gene Expression Omnibus(GEO)(GSE43458,GSE32863,and GSE27262)and the Cancer Genome Atlas(TCGA)datasets,including 698 LUAD and 172 healthy(or adjacent normal)lung tissue samples.Univariate regression and LASSO regression analyses were used to screen differentially expressed genes(DEGs)related to patient prognosis,and multivariate Cox regression analysis was applied to establish the risk score equation and construct the survival prognosis model.Receiver operating characteristic curve and Kaplan-Meier survival analyses with clinically independent prognostic parameters were performed to verify the predictive power of the model and further establish a prognostic nomogram.RESULTS A total of 380 DEGs were identified in LUAD tissues through GEO and TCGA datasets,and 5 DEGs(TCN1,CENPF,MAOB,CRTAC1 and PLEK2)were screened out by multivariate Cox regression analysis,indicating that the prognostic risk model could be used as an independent prognostic factor(Hazard ratio = 1.520,P < 0.001).Internal and external validation of the model confirmed that the prediction model had good sensitivity and specificity(Area under the curve = 0.754,0.737).Combining genetic models and clinical prognostic factors,nomograms can also predict overall survival more effectively.CONCLUSION A 5-mRNA-based model was constructed to predict the prognosis of lung adenocarcinoma,which may provide clinicians with reliable prognostic assessment tools and help clinical treatment decisions.
Key Words:Lung adenocarcinoma;Differentially expressed genes;Prognostic signature;Risk score;Nomogram
INTRODUCTION
Lung adenocarcinoma(LUAD)is a common histological type of lung cancer that is a malignant tumor that seriously threatens human health,accounting for approximately 40% of lung cancers[1].In recent years,some progress has been made in diagnostic and treatment strategies of clinical and experimental oncology for lung cancer.However,LUAD patients with localized or locally advanced disease still have a high risk of death,and their 5-year overall survival rate is still less than 15%[2].Assessing the patient's prognosis can help choose effective treatments to balance side effects with treatment benefits and decide whether to give more aggressive treatment.Although tumor-node-metastasis(TNM)classification plays an important role in the prognosis assessment of LUAD patients,the prognosis of some patients is significantly different even if the stages are similar.Therefore,the identification of reliable prognostic biomarkers to predict clinical outcomes and help make accurate clinical treatment decisions is clearly critical.The rapid development of gene chips and high-throughput sequencing have facilitated the development of new predictive tools based on prognostic genes for lung cancer.These relevant studies involved in prognostic genes of lung cancer have identified several prognostic models that have predicted the overall survival rate of LUAD patients(Table 1)[3-14].For example,a six-gene model(RRAGB,RSPH9,RPS6KL1,RXFP1,RTL1 and RRM2)based on the weighted gene coexpression network predicted the overall survival rate of non-small-cell lung cancer patients[13].A 3-gene prognostic model(URKA,CDC20,and TPX2A)also accurately predicted overall survival in smokingrelated lung adenocarcinoma[14].In addition,through analysis of TCGA data,the risk score of the 12-mRNA signature was correlated with poor prognosis in patients with lung adenocarcinoma[3].Therefore,the in-depth exploration of public databases such as the Gene Expression Omnibus(GEO)and the Cancer Genome Atlas(TCGA)databases,discovery of other genes related to the prognosis of LUAD and development of a comprehensive prognosis assessment system including multiple biomarkers may be effective ways to predict the prognosis of lung adenocarcinoma and individual treatment.
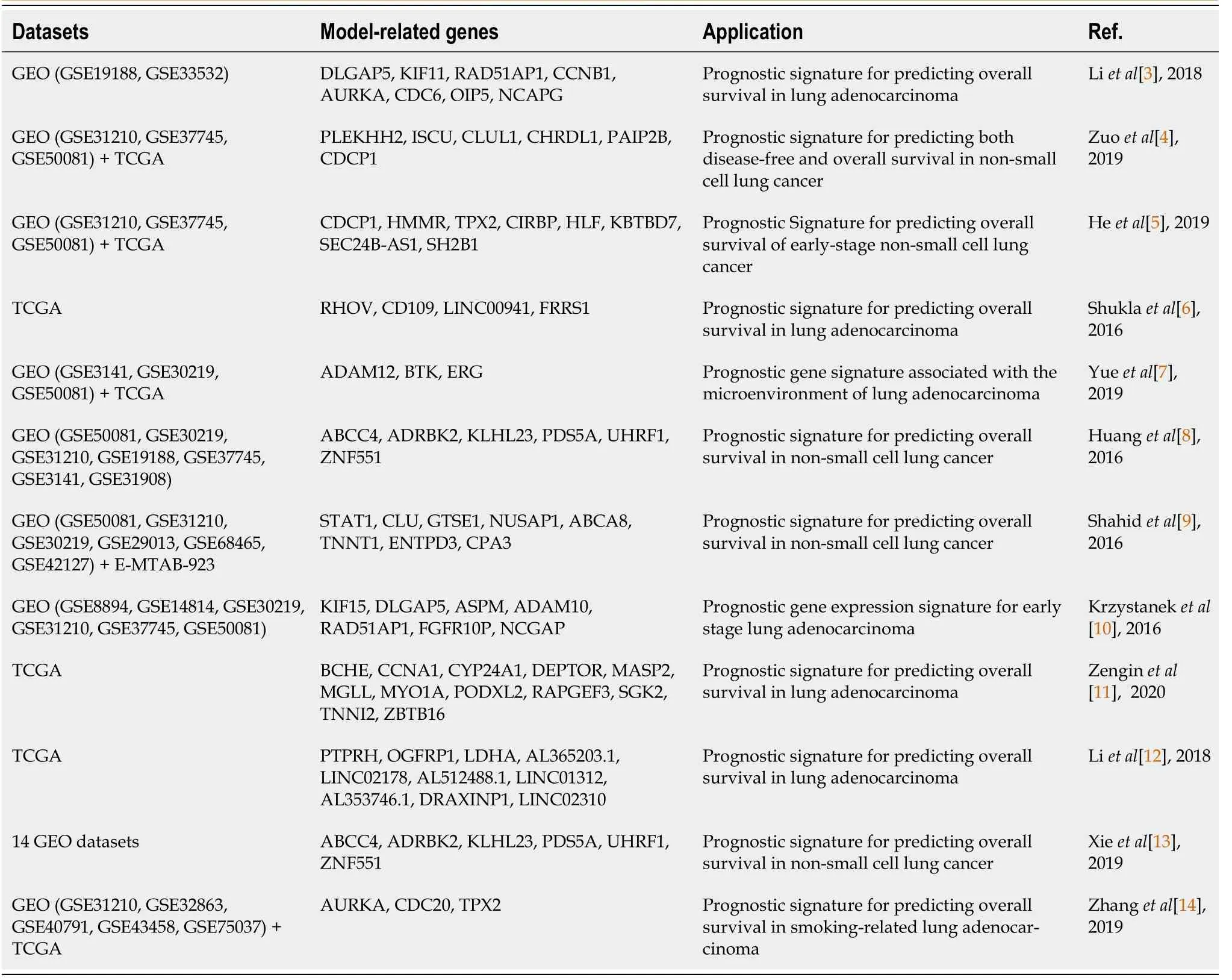
Table 1 Data and studies involved several prediction models for the prognostic signature of non-small cell lung cancer/ lung adenocarcinoma
Here,we first integrated three lung adenocarcinoma datasets from the GEO database to screen for differentially expressed genes(DEGs).Then,the TCGA-LUAD data set was used to identify DEGs.Univariate Cox and LASSO regression analyses were further used to determine the DEGs associated with overall survival.The risk score was calculated by multiplying multiple Cox coefficients by gene expression.The prognostic model was also combined with clinical parameters to construct a prognostic nomogram to predict overall survival.Finally,Gene set enrichment analysis(GSEA)was performed to identify the potential biological pathways of the five genes in the model.
MATERIALS AND METHODS
Gene expression profile data collection from the GEO and TCGA databases
The GEO database(https://www.ncbi.nlm.nih.gov/geo/)was used for the mRNA expression and clinical data of lung adenocarcinoma that needed to meet the following criteria:(1)Human lung adenocarcinoma tissue samples;(2)tumor and nontumor lung control tissue samples;and(3)≥ 50 samples.Finally,three gene expression microarray data sets(GSE43458,GSE32863 and GSE27262),which included 163 LUAD tumor tissue samples and 113 adjacent normal tissue samples,were downloaded for DEG analysis.On the other hand,the original count data and corresponding clinical data of LUAD patients in the training set and test set,which includes 535 LUAD patient samples and 59 control samples,were downloaded from TCGA project(https://tcga-data.nci.nih.gov/tcga/).Complete survival information and gene expression profile data of 494 patients were obtained from the TCGA database after excluding samples that could not be assessed for tumor histological grade or had no overall survival(OS)information.The model was validated using transcriptome analysis of 90 LUAD patients from the GSE11969 dataset.The workflow of our LUAD biomarker analysis process is shown in Supplementary Figure 1.
Screening and verification of DEGs in lung adenocarcinoma tissue
To identify DEGs between LUAD and lung tissues,GE02R was used for differential expression analysis of the GSE43458,GSE32863,and GSE27262 data sets.The DEGs of the TCGA-LUAD dataset were analyzed using the "limma" software package of R software,and the threshold of DEG screening was |log FC| > 2 andP< 0.05 according to our previous study[15].Human protein mapping(https://www.proteinatlas.org/)evaluates lung adenocarcinoma and DEG protein expression in normal lung tissue[16].Mutation data in lung adenocarcinoma patients were obtained from cBioPortal(https://www.cbioportal.org/)[17].
Identification of prognostic differential genes and establishment of prognostic models
TCGA-LUAD data were randomly divided into a training set(n= 346)and a test set(n= 148).In the test set,we performed univariate Cox regression analysis for DEGs determined by a comprehensive analysis of the GEO data set to determine the relationship between patient survival and gene expression.P< 0.01 was considered statistically significant and was included in subsequent analysis.Next,we applied LASSO regression to further reduce the number of DEGs in the selected panel with the best predictive performance by 10-fold cross-validation of the R-based glmnet package.Finally,multivariate Cox regression analysis was performed to obtain the five optimal prognostic gene regression coefficients from the multivariate Cox proportional hazard regression model.A prognostic risk score for the five genes was then established based on the multivariate Cox regression model regression coefficient multiplied by a linear combination of its mRNA expression level.
Identification of prognostic models and related genes
The Lung Adenocarcinoma(TCGA,PanCancer Atlas)database in cBioPortal was used to analyze the genetic mutation model.We used data from the TCGA to analyze model-related gene expression.The THPA(http://www.proteinatlas.org)database was used to analyze the protein expression of modelrelated genes[16].Patients in the training set were divided into high-risk and low-risk groups according to the median risk score as the cutoff point.Kaplan-Meier(KM)survival curves and Wilcoxon tests combined with the R package "survival" were used to compare the survival differences between the high-risk and low-risk groups.Time-dependent receiver operating characteristic(ROC)curve analysis was conducted using the R software package "survivalROC" to assess the prediction model's forecasting capacity.P< 0.05 was considered statistically significant.The test cohort and the entire cohort were used for internal validation,the GSE11969 dataset was downloaded from the GEO database for external validation,and the risk score of each patient was calculated using the same model based on the prognostic gene signature to further verify the predictive value of the prognostic gene signature.
Establish and verify the forecast nomograms
To provide clinicians with a quantitative method for predicting 1-year,3-year,and 5-year overall survival in LUAD patients,we used a combined model of all independent prognostic factors selected by multivariate Cox regression analysis to construct a nomogram.KM analysis,area under the curve(AUC),consistency index(C-index),and comparison of predicted and observed overall survival were used to evaluate the prognostic nomographs' performance[18].
Functional enrichment analysis of model genes
GSEA was used to analyze the signaling pathways of relevant genes involved in the development of lung adenocarcinoma to clarify the molecular mechanism of the prognostic gene signature.GSEA software(GSEA 4.0.3)was downloaded from the Broad Institute website(http://software.broadinstitute.org/gsea/index.jsp),and the analyzed access was from the c2.cp.kegg.v7.0.symbols.gmt data set in the Molecular Signature Database(MsigDB).The enrichment analysis was carried out by the weighted enrichment method,and the number of random combinations was set as 1000.All other parameters were set as default values.Gene sets withP< 0.05 were regarded as significantly enriched gene sets.
Statistical analysis
Statistical analysis and c orresponding graph drawing were performed using R3.6.3 software,and Cox regression analysis of the hazard ratio(HR)and 95% confidence interval(CI)was used to evaluate the association between DEG expression and prognosis.At-test of paired samples or a nonparametric Wilcoxon rank sum test of unpaired samples was used for analysis of continuous variables,and categorical variables were tested by the chi-square test or Fisher’s exact test.P< 0.05 indicated a significant difference.
RESULTS
Screening and identification of differentially expressed genes in the TCGA-LUAD database
We researched the results as described in the flowchart(Supplementary Figure 1).This study analyzed three GEO datasets(GSE43458,GSE32863,and GSE27262),and 886,1270,and 1921 DEGs were found,respectively.Then,we found 380 common DEGs by Venn diagram analysis.DEGs were verified in the TCGA-LUAD database(535 Lung adenocarcinoma tissues and 59 Lung cancer tissues),further confirming the differential expression of these 380 genes in lung adenocarcinoma and normal lung tissues(Figure 1A-D).
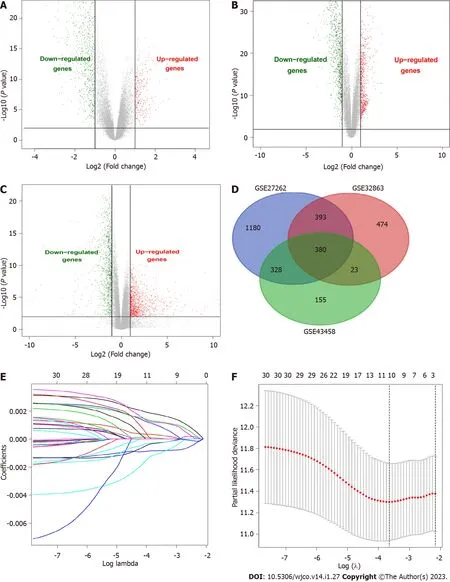
Figure 1 Screening of differential genes and establishing LASSO regression.A:Volcano map of the differential genes in the GSE43458;B:Volcano map of the differential genes in the GSE32863;C:Volcano map of the differential genes in the GSE27262;D:Venn diagram of the three Gene Expression Omnibus datasets;E:LASSO coefficients profiles of 380 common differential genes;F:LASSO regression with ten- fold cross-validation constructed the models.
Screening of prognostic differential genes and establishment of prognostic models
Univariate Cox regression analysis was performed on 380 DEGs in the training set.A total of 30 DEGs were related to the survival of patients with lung adenocarcinoma(P< 0.05)and further screened by LASSO regression(Figure 1E).Cross-validation was used to establish the model,as shown in Figure 1F.A total of 5 mRNAs(TCN1,CENPF,MAOB,CRTAC1,and PLEK2)were included in the model.Multivariate Cox regression analysis was performed for the above 5 mRNAs,and the risk scoring equation was established according to the corresponding regression coefficient.Risk score(RS)= (0.00288* TCN1 EXP)+(0.0387* CENPF EXP)+(-0.0291* MAOB EXP)+(-0.0198 *CRTAC1 EXP)+(0.0214* PLEK2 EXP).
Verification of mRNA expression and genetic changes associated with 5 prognostic genes
Among the 566 patients included in the cBioPortal for Lung Adenocarcinoma(TCGA,PanCancer Atlas)database,93 patients(16.4%)showed genetic changes in these 5 genes,among which missense mutations were the most common mutation type(Figure 2A).In the TCGA LUAD cohort,the mRNA expression levels of TCN1,CENPF,and PLEK2 were significantly increased in lung adenocarcinoma tissues compared with those in normal lung tissues,while MAOB and CRTAC1 were significantly decreased in lung adenocarcinoma tissues(Figure 2B).A human protein mapping database(http://www.proteinatlas.org)was used to explore the protein expression level.Immunohistochemical(IHC)results of four genes(TCN1 was not included in the database)in lung cancer and normal lung gland tissues are shown in Figure 2C.Consistent with the mRNA results,IHC results showed that CENPF and PLEK2 had significantly higher mean expression levels in lung adenocarcinoma tissue than in normal lung tissue.In contrast,the CRTAC1 expression level was higher in normal lung tissue than in lung adenocarcinoma tissue.MAOB showed no difference between normal and lung adenocarcinoma tissues(Figure 2C).
Evaluation of five-mRNA prognostic model
Each patient's risk score in the training group was calculated based on the above risk score function.The "SurvMiner" R software package was used to obtain the median critical point,and the patients were divided into a high-risk group and a low-risk group(Figure 3A).As the RS score increased,the patients' survival time was shortened,and the number of deaths increased significantly(Figure 3B).Figure 3C shows the heatmap of 5 prognostic genes in the high- and low-risk groups.The KM survival curve indicated a lower overall survival in the high-risk group than in the low-risk group(P< 0.001)(Figure 3D).To further verify the prognostic assessment model's accuracy,we used the R "survival ROC" package to draw the ROC curve(Figure 3E).The results showed that the AUC values of the risk score model for predicting the overall survival at 1,3 and 5 years in patients with lung adenocarcinoma were 0.711,0.668 and 0.728,respectively,indicating that the multigene model had a good predictive ability for the OS of patients with lung adenocarcinoma.Multiple Cox regression analysis showed that RS,along with patient age and stage,could be independent prognostic factors for lung adenocarcinoma patients(Figure 3F).
Internal and external validation of the five-mRNA prognostic signature
To verify the predictive value of the 5-mRNA prognostic signature,we used the same formula to calculate risk scores for patients with the internal validation set(n= 160),entire validation set(n= 535),and external validation set(GSE11969,n= 90).Consistent with the training group results,the OS of LUAD patients in the high-risk group was significantly lower than that in the low-risk group(Figure 4A-C).The KM survival analysis of the prognostic signature showed that the AUC values of the 1-year,3-year,and 5-year OS of the internal validation set,the overall validation set and external validation set were 0.754,0.630,0.684,and 0.737,0.701,0.680,and 0.779,0.752,0.715,respectively(Figure 4D-F).Taken together,our results suggest that this 5-gene signature performs well in predicting overall survival in patients with lung adenocarcinoma.
Establish and verify the nomogram
To establish clinically applicable methods for predicting survival in patients with lung adenocarcinoma,we established a nomogram using three independent prognostic factors(including age,stage,and risk score)to predict 1-year,3-year,and 5-year OS in patients with lung adenocarcinoma(Supplementary Figure 2A).The calibration diagram showed that the nomogram performs well(Supplementary Figure 2B).The AUC values of the 1-year,2-year,and 3-year overall survival predictions of the nomograph were 0.760,0.712,and 0.709,respectively(Supplementary Figure 3A).The KM chart effectively distinguishes the various risks of these categories,with people with higher scores having significantly poorer overall survival(P< 0.001)(Supplementary Figure 3B).The C-index(95%CI)of the age,stage,and risk score and combination models was 0.501(0.480-0.522),0.684(0.662-0.076),0.625(0.604-0.646),and 0.726(0.702-0.750),respectively(Supplementary Table 1).Thus,the nomogram performs well in predicting overall survival in patients with lung adenocarcinoma,which may be useful for patient counseling and clinical decision-making.
Biological pathways of the five prognostic model genes were identified
GSEA was performed to identify the potential biological processes of the 5 prognostic genes and showed that the samples with highly expressed TCN1,CENPF,and PLEK2 were enriched with focal adhesion,the p53 signaling pathway,and Toll-like sensors,respectively.MAOB and CRTAC1 samples were mediated in the cell cycle and ubiquitin-mediated proteolysis(Supplementary Figure 4),respectively.
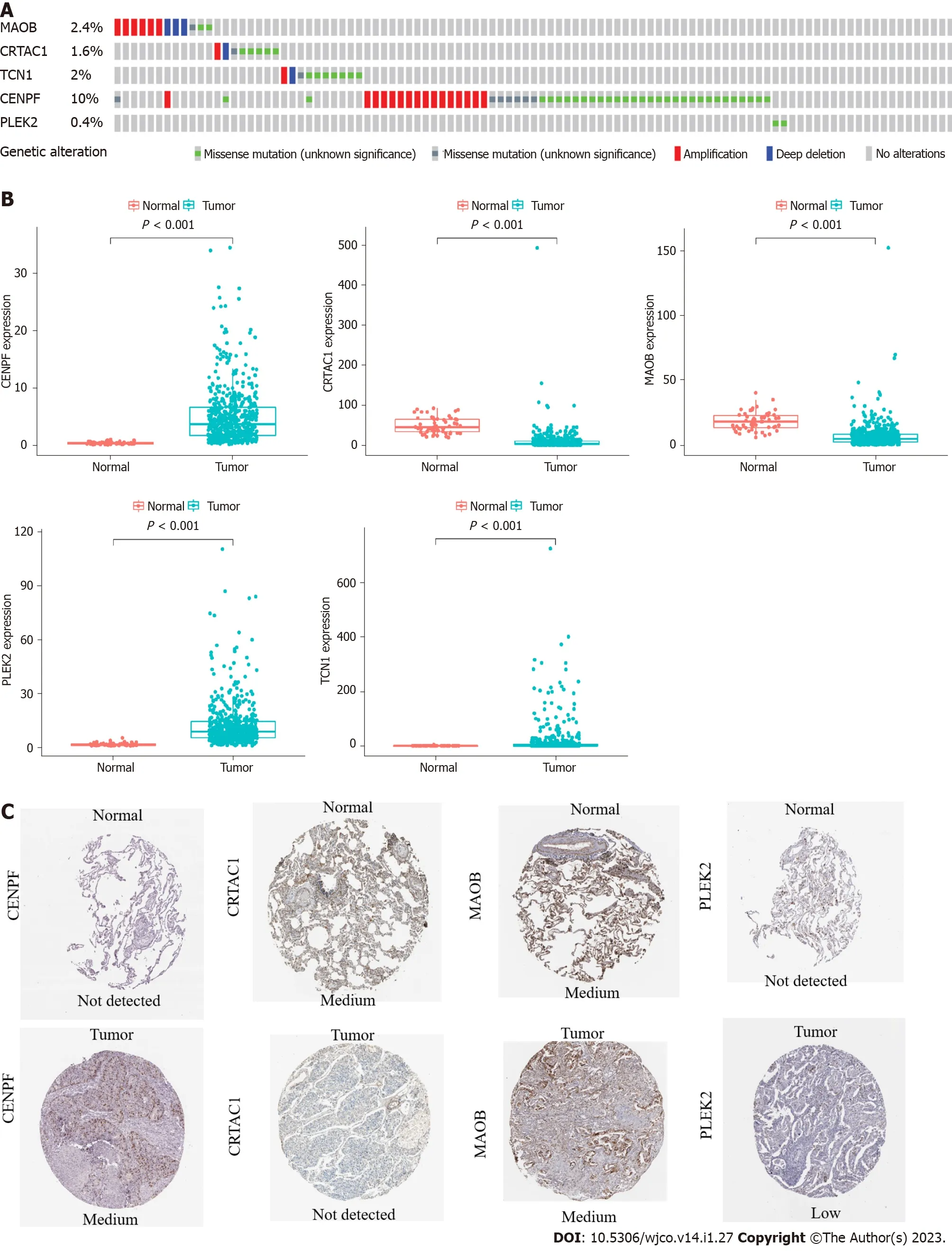
Figure 2 The expression and genetic alterations of the 5 prognostic genes in Lung adenocarcinoma.A:Genetic alterations rate of 5 model genes;B:Differential expression of the mRNA levels in lung adenocarcinoma tissues;C:Differential expression at the protein levels of the five model genes.
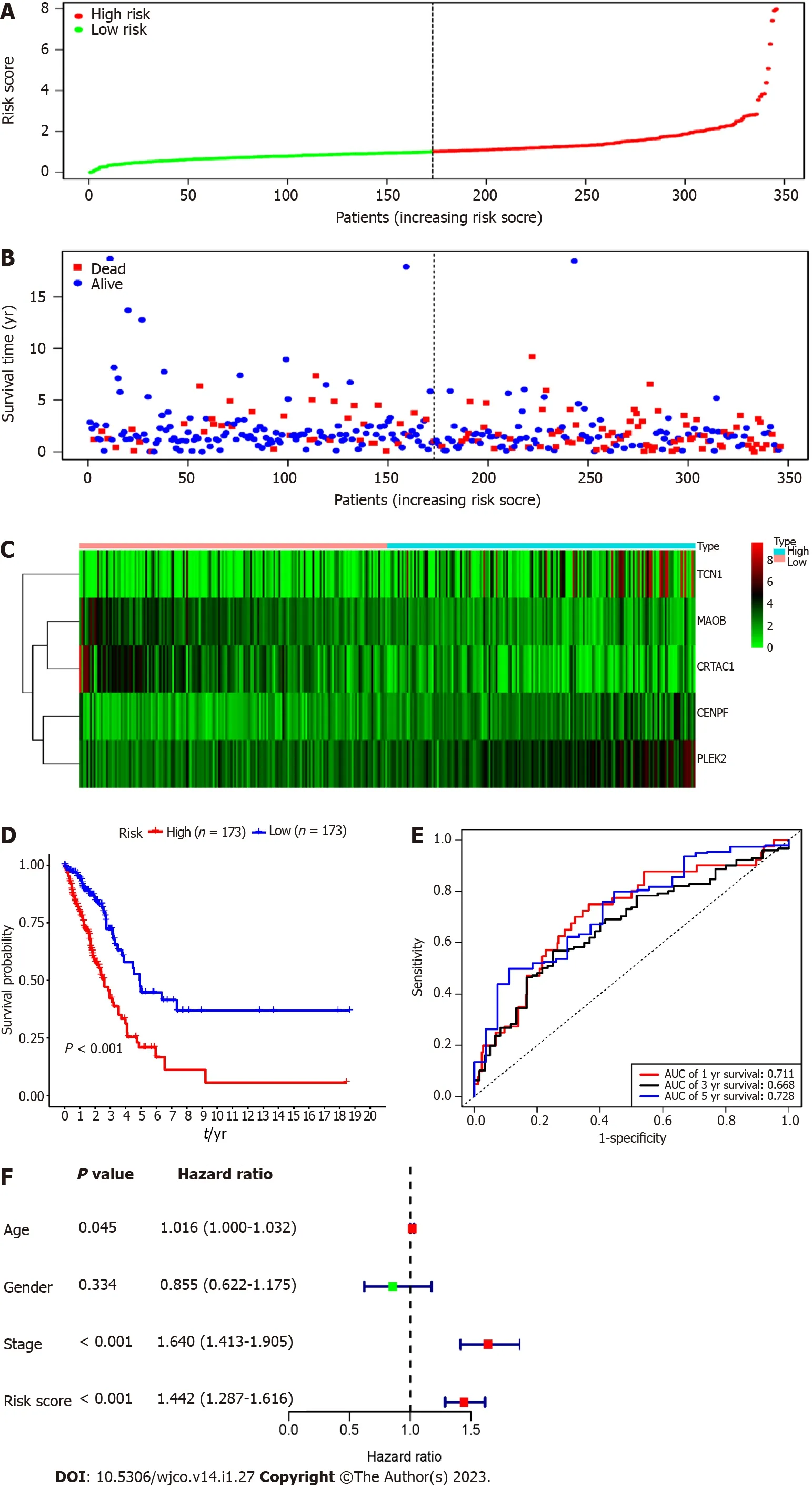
Figure 3 Risk score model,time-dependent receiver operating characteristic analysis,and survival analysis for the 5-gene signature in Lung adenocarcinoma.A:The risk score curve divided the patients into the high-risk and low-risk groups;B:Distribution map of the patient's survival status;C:Heatmap of model genes in high and low risk groups;D:Kaplan-Meier survival analysis of the 5-mRNA-based prognostic signature;E:Receiver operating characteristic curves to evaluate the prognostic signature;F:Multiple cox regression analysis of the risk scores and clinical parameters.
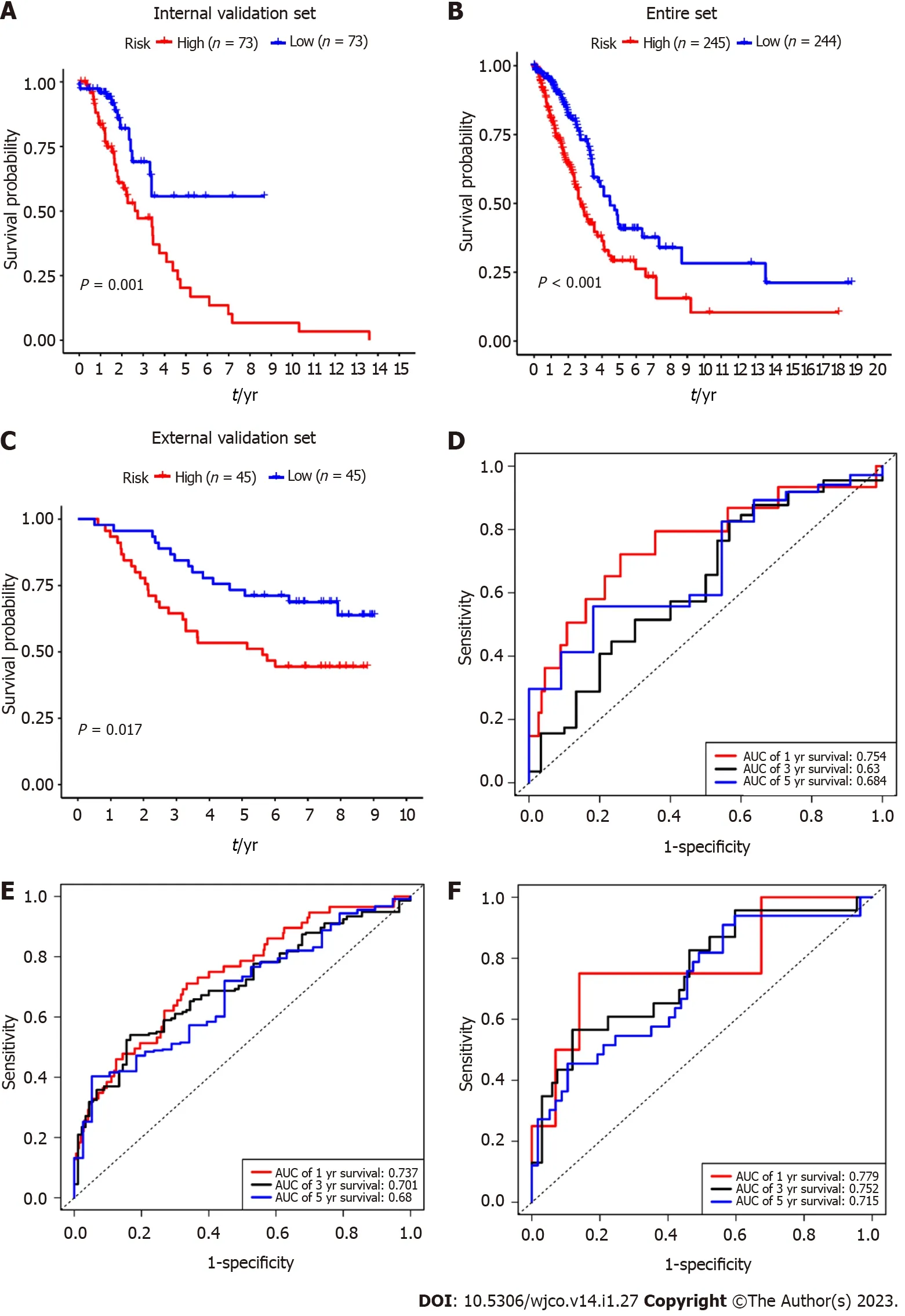
Figure 4 Internal and external validation of the 5-gene prognostic signature.A:Internal validation by survival analysis;B:The entire dataset validation by survival analysis;C:GSE11969-based external validation by survival analysis;D:Internal validation by receiver operating characteristic(ROC)curves;E:The entire dataset validation by ROC curves;F:GSE11969-based external validation by ROC curves.
DISCUSSION
Recently,the tumor prognosis model based on the abnormal gene mRNAs has shown great potential due to its high prediction accuracy.Traditional clinicopathological parameters,such as tumor stage,have been used to reflect and predict disease progression.However,a single clinical parameter has poor predictive ability for prognosis[3,6,7,10].In this study,we identified 380 reliable lung adenocarcinoma differential genes by comprehensive analysis of three GEO datasets combined with data from the TCGA-LUAD.Univariate,LASSO and multivariate Cox analyses of DEGs were performed to establish a prognostic risk model for lung adenocarcinoma based on 5 mRNAs(TCN1,CENPF,MAOB,CRTAC1,and PLEK2).These five new genes were significantly correlated with the prognosis of LUAD patients.MAOB and CRTAC1 were negative prognostic genes,while TCN1,CENPF and PLEK2 were positive prognostic genes.Recently,several studies have revealed the important role of these five genes in cancer progression.Monoamine oxidase B(MAOB)is an enzyme located on the outer mitochondrial membrane.It is responsible for catalyzing monoamine oxidation to produce hydrogen peroxide and is mainly involved in the metabolism of neurotransmitters[19].The relationship between MAOB and tumors is less reported.It has been reported that MAOB mRNA is significantly lower in the saliva of oral squamous cell carcinoma patients than in that of healthy controls[20].Xuet al[21]found that MAOB is a key DNA methylation driver gene for prostate cancer and plays an important role in the DNA methylation of prostate cancer patients through a comprehensive analysis of the TCGA methylation data.There is no previous report about MAOB in lung adenocarcinoma.CRTAC1 encodes human chondrogenic acid protein 1,which can be used as a marker of chondrocytes to distinguish human chondrocytes from osteoblasts and mesenchymal stem cells in cultures[22].Currently,this gene is rarely reported in tumors.TCN1 is a member of the vitamin B12-binding protein family and is a 60-70 kDa molecular weight protein.High levels of TCN1 are primarily related to abnormal granulocyte proliferation.TCN1 is overexpressed in a variety of malignancies,such as pancreas,breast,and colon cancer,and is associated with tumor progression and metastasis[23-25].TCN1 was significantly associated with advanced colorectal cancer[24]and laryngeal cancer[26].Centromere protein F(CENPF),as an important member of the centromere protein family,is a component of the centromere complex and plays an important regulatory role in mitosis[27].CENPF expression is abnormally increased in a variety of malignant tumors and is associated with the prognosis of patients[28,29].Using bioinformatics and immunohistochemical analysis,CENPF overexpression was associated with poor prognosis of breast cancer and tumor bone metastasis[30].Through comprehensive analysis of three GEO databases,CENPF was identified as a key gene with prognostic value in lung adenocarcinoma,which was consistent with our research results[31].Pleckstrin-2(PLEK2)is a 353 amino acid protein encoded by the PLEK2 gene in the human genome and is widely expressed in various tissues.Its overexpression contributes to the formation of large apolipoproteins,thereby promoting cell proliferation[32].PLEK2 has been found to be related to the invasion and metastasis of multiple tumors.In gallbladder cancer(GBC),PLEK2 overexpression enhances the epithelial-mesenchymal transformation(EMT)process in GBC cells,leading to subsequent higher rates of cell migration,invasion,and liver metastasis[33].The overexpression of PLEK2 also significantly promoted the EMT and migration of non-small cell lung cancer and destroyed the vascular endothelial barrier[34].After identifying the five prognostic gene markers,we also conducted internal and external validation to confirm their predictive value and revealed that the prognostic signatures had good prognostic diagnostic value.To improve the prognostic predictive power of the five prognostic gene markers,a predictive nomogram combining risk scores and conventional clinical prognostic parameters(including age and tumor stage)was constructed to enable clinicians to determine the prognosis of each patient.Its graphical scoring system is easy to understand and helps customize treatment and medical decisions.The prognostic models and nomograms associated with five-gene characteristics have not been reported.Hence,our study may be useful prognostic and diagnostic classification tools for lung adenocarcinoma.Our study still has some limitations.First,the study only focuses on transcriptome sequencing data.If other omics techniques,such as DNA methylation and single nucleotide polymorphisms,can be analyzed together,more favorable results may be obtained.Second,our research is limited to the bioinformatics analysis of the TCGA and GEO databases.Although we have verified the accuracy of the models internally and externally,the verification of large samples in the clinical diagnosis and treatment process will further enhance their diagnostic accuracy and clinical value.
CONCLUSION
In summary,our study identified a 5-gene model and prognostic nomogram that combined gene models and clinical prognostic factors to predict the overall survival rate of lung adenocarcinoma patients,and this nomogram may be of great significance for the selection of personalized treatment options and clinical medical decisions in patients with lung adenocarcinoma.
ARTICLE HIGHLIGHTS
Research background
Lung adenocarcinoma patients with localized or locally advanced disease have a high risk of death,and their 5-year overall survival rate is less than 15%.
Research motivation
To evaluate the prognosis of Lung adenocarcinoma(LUAD)patients and optimize treatment,effective clinical research prediction models.
Research objectives
To identify reliable prognostic biomarkers to predict clinical outcomes and to help clinicians to make accurate clinical treatment decisions.
Research methods
The Cancer Genome Atlas(TCGA)and the Gene Expression Omnibus(GEO)were used to screen for differential genes for lung adenocarcinoma.Univariate regression analysis combined with LASSO regression analysis was used to screen for prognostic-related genes.Multivariate Cox regression analysis was applied to establish the risk score equation and construct the survival prognosis model.
Research results
We establish a prognostic risk model for lung adenocarcinoma based on 5 mRNAs(TCN1,CENPF,MAOB,CRTAC1,and PLEK2).These five new genes were significantly correlated with the prognosis of LUAD patients.To improve the prognostic predictive power of the five prognostic gene markers,a predictive nomogram combining risk scores and conventional clinical prognostic parameters(including age and tumor stage)was constructed to enable clinicians to determine the prognosis of each patient.
Research conclusions
A 5-mRNA-based model was constructed to predict the prognosis of lung adenocarcinoma,which may provide clinicians with reliable prognostic assessment tools and help clinical treatment decisions.
Research perspectives
Our study identified a 5-gene model and constructed a nomogram which may have important implications for clinical medical decision and personalized treatment of patients with lung adenocarcinoma.
FOOTNOTES
Author contributions:Wang J,Du YZ,and Xia QL conceived and designed the experiments;Xia QL,He XM and Ma Y analyzed the data;Li QY contributed to analysis tools;Xia QL wrote the manuscript;Wang J and Xia QL revised the manuscript;all authors have read and approved the final manuscript.
Supported bythe Science and Technology Development Fund of the Pudong New Area,No.PKJ2021-Y53;and the National Natural Science Foundation of China,No.81974315.
Institutional review board statement:The study was reviewed and approved by the Shanghai Public Health Clinical Center Laboratory Animal Welfare &Ethics Committee Institutional Review Board[(Approval No.2020-A006-01]).
Conflict-of-interest statement:All the authors declare no competing financial interests.
Data sharing statement:The mRNA expression and clinical data of lung adenocarcinoma analyzed during the current study are available on the GEO(https://www.ncbi.nlm.nih.gov/geo/)and TCGA databases(https://www.cbioportal.org/).The protein expression of model-related genes of LUAD analyzed in this study is also available on the THPA database(http://www.proteinatlas.org).
Open-Access:This article is an open-access article that was selected by an in-house editor and fully peer-reviewed by external reviewers.It is distributed in accordance with the Creative Commons Attribution NonCommercial(CC BYNC 4.0)license,which permits others to distribute,remix,adapt,build upon this work non-commercially,and license their derivative works on different terms,provided the original work is properly cited and the use is noncommercial.See:https://creativecommons.org/Licenses/by-nc/4.0/
Country/Territory of origin:China
ORCID number:Yu-Zhen Du 0000-0002-3395-4471;Jin Wang 0000-0002-0062-2489.
S-Editor:Liu JH
L-Editor:A
P-Editor:Liu JH
