
HSP90抑制剂前药设计及合成
2023-03-15 17:00刘文金曹小冬
浙江理工大学学报 2023年11期
关键词:酰化
刘文金 曹小冬
摘 要: 为解决热休克蛋白90(Heat shock protein 90,HSP90)抑制剂的周身毒性问题,自主设计并合成了两个HSP90抑制剂的三肽前药P1和P2。以4-(2-羟乙基)哌啶-1-甲酸叔丁酯和1-(溴甲基)-4-硝基苯为起始原料,经羟胺缩合、硝基还原、关环等反应合成HSP90抑制剂HSP90i-1和HSP90i-2;以L-脯氨酸和L-缬氨酸衍生物为原料,经缩合、脱保护、酰化等反应合成多肽连接子M1。将HSP90i-1、HSP90i-2分别和M1缩合,得到两个三肽前药P1和P2。1H NMR、LCMS、13C NMR分析表征结果表明成功合成了两个前药分子两个前药分子被成功合成。该研究具有使用的原料廉价易得、反应都在常温下进行、条件温和可控、总产率较高等特点,为多肽前药的设计及合成提供了新的思路和方法。
关键词: HSP90抑制剂;前药;缩合;酰化;多肽前药
中图分类号: O622.6
文献标志码: A
文章编号: 1673-3851 (2023) 09-0604-08
引文格式:刘文金,曹小冬. HSP90抑制剂前药设计及合成[J]. 浙江理工大学学报(自然科学),2023,49(5):604-611.
Reference Format: LIU Wenjin, CAO Xiaodong. Design and synthesis of HSP90 inhibitor prodrugs[J]. Journal of Zhejiang Sci-Tech University,2023,49(5):604-611.
Design and synthesis of HSP90 inhibitor prodrugs
LIU Wenjin, CAO Xiaodong
(School of Science, Zhejiang Sci-Tech University, Hangzhou 310018, China)
Abstract: To solve the peripheral toxicity problem of heat shock protein 90 (HSP90) inhibitors, we designed and synthesized two HSP90 inhibitors, P1 and P2, as tripeptide prodrugs. The HSP90 inhibitors HSP90i-1 and HSP90i-2 were synthesized from tert-Butyl 4-(2-hydroxyethyl) piperidine-1-carboxylate and 1-Bromo-4-nitrobenzene by the reactions of hydroxylamine condensation, nitroreduction, ring closure, etc. The peptide linker M1 was synthesized from L-proline and L-valine derivatives by the reactions of condensation, deprotection, acylation, etc. HSP90i-1 and HSP90i-2 were condensed with M1 to obtain the two tripeptide prodrugs of P1 and P2.1H NMR, LCMS,13C NMR analysis and characterization results indicated that the two prodrugs were successfully synthesized. The study is characterized by the availability of inexpensive raw materials, the mild and controlled conditions of the reactions at room temperature, and the high overall yields, which provides new ideas and methods for the design and synthesis of polypeptide prodrugs.
Key words: HSP90 inhibitor; prodrug; condensation; acylation; polypeptide prodrug
0 引 言
热休克蛋白90(Heat shock protein 90,HSP90)是一种癌症治疗靶点,近年来备受关注。HSP90通过与客户蛋白和辅助分子伴侣的相互作用调节细胞内的信号通路、蛋白质稳态和细胞凋亡过程。通常,HSP90可用于保持400多个蛋白的构象稳定,同时也是癌细胞生长和转移的关键调节因子,能促进瘤细胞运动、侵袭和转移[1]。HSP90治疗靶点发现至今已有三十多年时间,尽管有三十多个HSP90抑制剂进入了临床研究,但都因安全性问题宣告失败[2-3]。Synta公司研发的Ganetespib,在前期研究中对非小细胞肺癌表现出良好的治疗效果,却在临床三期试验中因毒性而終止后续研究[4]。前药策略是将母药分子进行化学结构修饰,使活性母药与某些功能基团以共价键相连,在体内经由某些生物酶的作用释放出母药,从而达到较好的药效。前药策略在药物开发过程中被广泛应用,如用于提高口服药物的生物利用度、增强药物的血脑屏障渗透性、增强药物化学代谢以及母药的靶向递送等[5-8]。将前药策略用于母药的靶向递送,在抗肿瘤药物开发领域极具前景,能够极大地提高药物的安全性。
HSP90抑制剂的毒性主要来源于两个方面:一方面是由于药物进入体内循环后,在各个器官广泛分布,对正常组织的HSP90蛋白功能产生抑制作用,从而诱发周身毒性;另外,进入临床的HSP90抑制剂的选择性过大,尤其对HSP90蛋白家族的其他亚型也都有较强的抑制作用,不可避免地产生脱靶毒性。因此,HSP90亚型选择性抑制剂的开发已经成为一个重要的研究方向。然而,HSP90的蛋白结构高度保守,这使得开发选择性抑制剂异常困难[9-14]。
本文以4-(2-羟乙基)哌啶-1-甲酸叔丁酯和1-(溴甲基)-4-硝基苯为起始原料,经缩合、还原、关环等多步反应得到两个HSP90抑制剂HSP90i-1和HSP90i-2;以L-脯氨酸和L-缬氨酸衍生物为原料,经过缩合、脱保护、酰化反应得到三肽连接子M1。将M1分别与HSP90i-1和HSP90i-2缩合,得到三肽前药分子P1和P2。利用1H NMR、LCMS、13C NMR等分析中间体及目标分子的结构。本文的研究可为三肽类前药提供新的设计思路和合成方法。
1 实验部分
1.1 实验材料及仪器
实验材料:N,N-二甲基甲酰胺(DMF)、二氯甲烷(DCM)、四氢呋喃(THF)、甲醇(MeOH)、乙醇(EtOH),三氟乙酸(TFA)均为分析纯,购于上海安耐吉化学有限公司;4-二甲氨基吡啶(DMAP)、盐酸二恶烷(HCl)、1-羟基-7-氮杂苯并三氮唑(HOAT)、2-(7-氮杂苯并三氮唑)-N,N,N′,N′-四甲基脲六氟磷酸酯(HATU)、2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉(EEDQ)、对氨基苄醇(PAB)、N -乙基二异丙胺(DIEA)、三乙胺(TEA)、对硝基氯甲酸苯酯、对甲苯磺酰氯(TsCl)、氢化钠(NaH)、钯碳(Pd/C)、碳酸钠(Na2CO3)、羰基二咪唑(CDI)、水合肼(NH2-NH2·H2O),均为分析纯,购于上海毕得医药。
实验仪器:Bruker Avance AV(400 MHz)核磁共振波谱仪,Biotage LCQ-Fleet质谱仪,waters Biotage Isolera Prime快速制备液相色谱,岛津 UV-2600紫外分光光度仪。
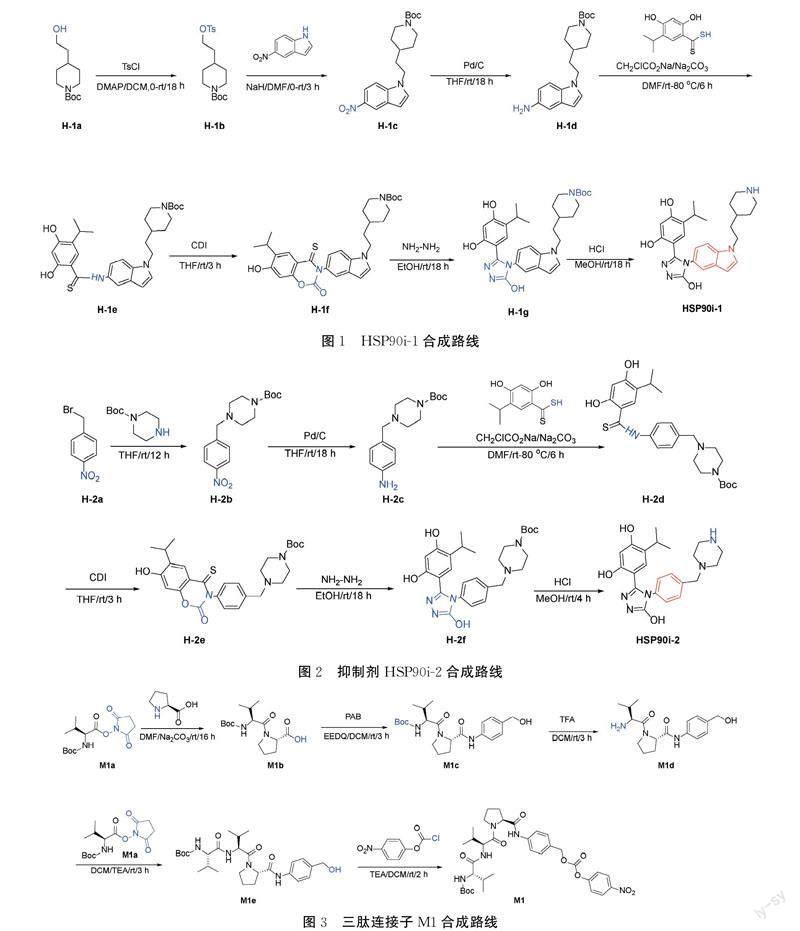
1.2 抑制剂HSP90i-1合成
目标分子的合成路线见图1。4-(2-羟乙基)哌啶-1-甲酸叔丁酯经TsCl活化羟基、羟胺缩合、硝基还原、关环和脱保护[15]等步骤合成4-(5-羟基-4-(1-(2-(哌啶-4-基)乙基)-1H-吲哚-5-基)-4H-1,2,4-三唑-3-基)-6-异丙基苯-1,3-二醇(HSP90i-1)。1H NMR (400 MHz, DMSO) δ 11.90 (s, 1H), 9.07 (d, J=8.7 Hz, 1H), 8.88 (d, J=9.4 Hz, 1H), 7.50~7.45 (m, 2H), 7.43 (d, J=1.9 Hz, 1H), 6.93 (dd, J=8.7, 1.9 Hz, 1H), 6.69 (s, 1H), 6.43 (d, J=3.0 Hz, 1H), 6.32 (s, 1H), 4.21 (t, J=7.1 Hz, 2H), 3.22~3.16 (m, 2H), 2.89 (m, 1H), 2.75 (q, J=11.7 Hz, 2H), 1.83 (d, J=12.6 Hz, 2H), 1.68 (q, J=13.5, 6.7 Hz, 2H), 1.49~1.32 (m, 3H), 0.81 (d, J=6.9 Hz, 6H)。LCMS[M+H]+=462.3。
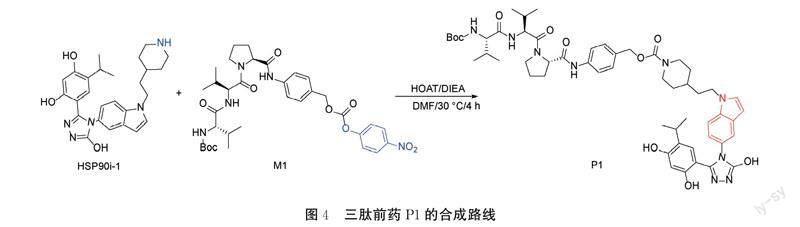
1.3 抑制剂HSP90i-2的合成
目标分子的合成路线见图2。将1-(溴甲基)-4-硝基苯经取代、还原、缩合、关环和脱保护等步骤合成4-(5-羟基-4-(4-(哌嗪-1-基甲基)苯基)-4H-1,2,4-三唑-3-基)-6-异丙基苯-1,3-二醇(HSP90i-2)。1H NMR (401 MHz, DMSO) δ 11.92 (s, 1H), 9.51 (d, J=82.9 Hz, 2H), 7.73~5.65 (m, 6H), 3.46 (s, 2H), 3.29 (s, 4H), 3.06~2.87 (m, 1H), 2.62~2.18 (m, 43H), 1.38 (s, 9H), 0.94 (d, J=6.9 Hz, 6H)。LCMS[M+H]+=410.1。
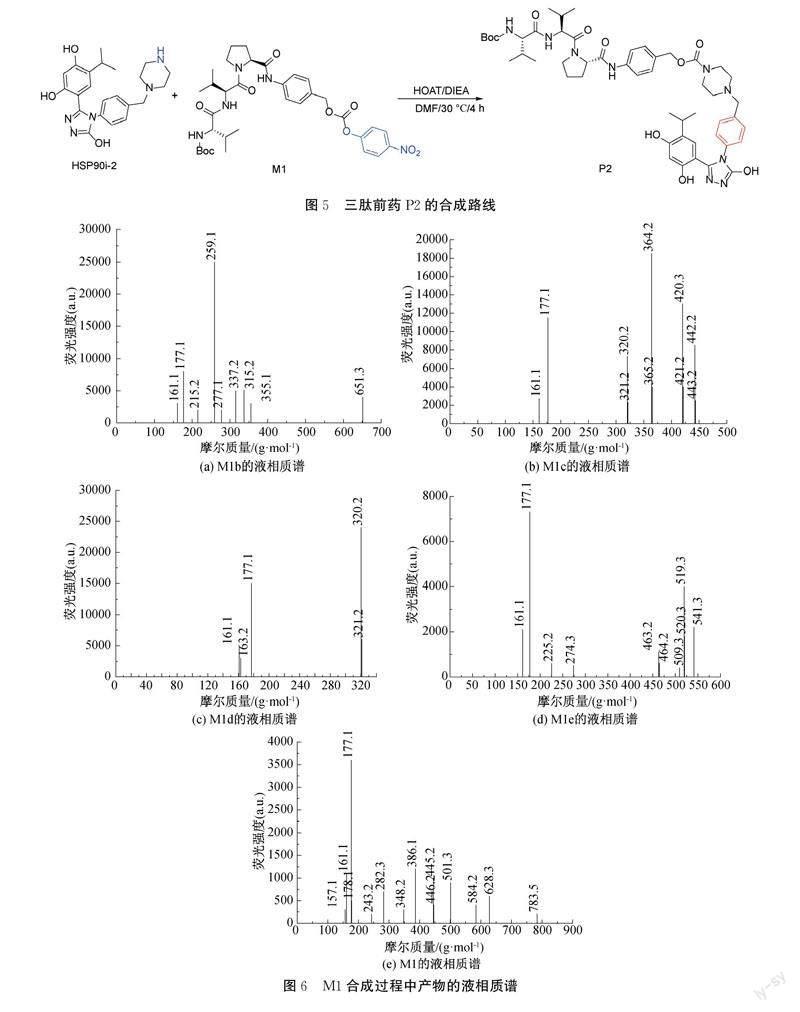
1.4 三肽连接子M1合成
目标分子的合成需经历5步反应,具体合成路线见图3。
1.4.1 M1b合成
将L-缬氨酸衍生物(M1a,0.9 g, 2.95 mmol)溶于DMF(20 mL),加入脯氨酸(0.3 g, 2.95 mmol)和碳酸钠(0.5 g, 4.43 mmol),室温反应16 h。加水(10 mL),用乙酸乙酯(50 mL×4)萃取,无水硫酸钠干燥有机层,减压浓缩,得白色固体(672.0 mg, 收率:75%)。LCMS[M+H]+=259.1。
1.4.2 M1c合成
将M1b(0.5 g, 1.59 mmol)和PAB(0.2 g, 1.75 mmol)加入二氯甲烷(20 mL),加入EEDQ(0.8 g, 3.18 mmol),室温反应3 h。减压浓缩,硅胶色谱法纯化(PE/EtOAc=7/3PE与EtOAc比值为7∶3)得白色固体(513.0 mg, 收率:77%)。LCMS[M+H]+=420.2。
1.4.3 M1d合成
將M1c(0.6 g, 1.43 mmol)加入二氯甲烷(20 mL),加入三氟乙酸(0.4 mL, 4.30 mmol),室温反应3 h。减压浓缩,硅胶色谱法纯化(PE/EtOAc=4/1PE与EtOAc比值为4∶1)得白色固体(379.0 mg, 收率:80%)。LCMS[M+H]+=320.2。
1.4.4 M1e合成
将M1d(0.1 g, 1.30 mmol)溶于二氯甲烷(10 mL),加入M1a(0.2 g, 1.40 mmol)和三乙胺(0.3 mL, 1.95 mmol),室温反应3 h。加水(5 mL),用乙酸乙酯(20 mL×4)萃取,无水硫酸钠干燥有机层,减压浓缩,得白色固体(110 mg, 收率:69%)。1H NMR (401 MHz, DMSO) δ 10.15~9.67 (m, 1H), 7.77 (d, J=8.3 Hz, 1H), 7.66~7.41 (m, 2H), 7.23 (d, J=8.5 Hz, 2H), 6.84 (dd, J=23.6, 9.2 Hz, 1H), 5.76 (s, 2H), 5.09 (t, J=5.6 Hz, 1H), 4.42 (d, J=5.2 Hz, 4H), 3.92~3.71 (m, 2H), 3.63 (d, J=6.7 Hz, 1H), 2.12 (d, J=8.5 Hz, 1H), 2.06~1.79 (m, 5H), 1.38 (s, 11H), 0.97~0.69 (m, 13H)。LCMS[M+H]+=519.3。
1.4.5 M1合成
将M1e(0.4 g, 1.30 mmol)溶于二氯甲烷(10 mL),加入对硝基氯甲酸苯酯(39.0 mg, 0.19 mmol),加入三乙胺(1.0 mL, 0.38 mmol),室温反应2 h。加水(5 mL),用乙酸乙酯(20 mL×4)萃取,无水硫酸钠干燥有机层,减压浓缩,得白色固体(441.9 mg, 收率:84%)。LCMS[M+H]+=628.3。
1.5 三肽前药P1的合成
目标分子的合成路线见图4。将HSP90i-1(80.0 mg, 0.17 mmol)溶于DMF(10 mL),加入M1(118.0 mg, 0.17 mmol)和HOAT(34.7 mg, 0.23 mmol),加入DIEA(41.0 mg, 0.34 mmol),在30 ℃反应4 h。减压浓缩,硅胶色谱法纯化(PE/EtOAc=7/3PE与EtOAc比值为7∶3)得白色固体(123.8 mg, 收率:71%)。1H NMR (401 MHz, DMSO) δ 11.87 (s, 1H), 10.06 (s, 1H), 9.52 (d, J=16.2 Hz, 2H), 7.77 (d, J=8.8 Hz, 1H), 7.47 (ddd, J=23.6, 14.1, 5.2 Hz, 5H), 7.28 (d, J=8.6 Hz, 2H), 6.93 (dd, J=8.6, 2.0 Hz, 1H), 6.81 (d, J=9.5 Hz, 1H), 6.68 (s, 1H), 6.42 (d, J=3.0 Hz, 1H), 6.23 (s, 1H), 4.98 (s, 2H), 4.38 (dd, J=15.9, 7.2 Hz, 2H), 4.20 (s, 2H), 3.95 (d, J=11.1 Hz, 2H), 3.82 (s, 2H), 3.63 (s, 1H), 2.95~2.77 (m, 1H), 2.10~1.79 (m, 6H), 1.67 (d, J=6.5 Hz, 4H), 1.38 (s, 11H), 1.17~0.99 (m, 3H), 0.98~0.85 (m, 7H), 0.80 (dd, J=12.7, 6.9 Hz, 12H)。LCMS[M+H]+=963.5。
1.6 三肽前药P2的合成
目标分子的合成路线见图5。将HSP90i-2(100.0 mg, 0.24 mmol)溶于DMF(10 mL),加入M1(167.0 mg, 0.24 mmol)和HOAT(49.0 mg, 0.36 mmol),加入DIEA(57.9 mg, 0.48 mmol),在30℃反应4 h。减压浓缩,硅胶色谱法纯化(PE/EtOAc=7/3PE与EtOAc比值为7∶3)得白色固体(158.5 mg, 收率:68%)。1H NMR (401 MHz, DMSO) δ 11.91 (s, 1H), 10.10 (d, J=23.2 Hz, 1H), 9.58 (s, 1H), 9.38 (s, 1H), 7.77 (d, J=8.9 Hz, 1H), 7.56 (d, J=8.5 Hz, 2H), 7.29 (dd, J=8.4, 4.6 Hz, 4H), 7.13 (d, J=8.3 Hz, 2H), 6.89~6.66 (m, 2H), 6.25 (s, 1H), 4.99 (s, 2H), 4.39 (dt, J=17.0, 8.3 Hz, 2H), 3.82 (s, 2H), 3.64 (s, 1H), 3.46 (s, 3H), 3.07~2.85 (m, 1H), 2.76~2.59 (m, 2H), 2.38-2.23 (m, 7H), 2.20~1.74 (m, 6H), 1.38 (s, 9H), 1.03~0.69 (m, 18H)。LCMS[M+H]+=954.2。
2 結果与讨论
2.1 M1的合成过程分析
对M1进行质谱分析测试,结果如图6所示。在三肽连接子M1的合成过程中,M1a和脯氨酸在DMF和Na2CO3条件下进行缩合,得到M1b。该氨基酸缩合反应条件温和,特异性高[16],收率为75%。图6(a)展示了M1b在液相质谱ESI正模式下测试所得的谱图;由图6(a)可知其中:横坐标为化合物的摩尔质量,单位为(g/mol),表示化合物的分子质量;纵坐标为荧光强度,用a.u.表示,表示化合物吸收强度。测得离子峰[M+H]+=259.1,因结构中包含Boc,由此推断所测样品中化合物分子量为259.1+56,与目标产物M1b的理论分子质量314.1一致。
由于无保护的二肽很容易发生自身缩合,形成稳定的哌嗪二酮结构[17],故在引入第三个氨基酸之前,先与M1b的羧基反应,即M1b和对氨基苄醇反应得到二肽的苯酰胺衍生物M1c,纯化后产物收率为77%。图6(b)展示了M1c在液相质谱ESI正模式下测试所得的谱图;由图6(b)可知,测得离子峰[M+H]+=420.3,与目标产物M1c的理论分子质量419.3一致。
M1c在DCM中用TFA脱去氨基保护基Boc,得到N端未保护的二肽M1d,收率80%。图6(c)展示了M1d在液相质谱ESI正模式下测试所得的谱图;由图6(c)可知,测得离子峰[M+H]+=320.2,与目标产物M1d的理论分子質量319.2一致。
M1d中的氨基与M1a的活性酯部分高效缩合得到三肽苯酰胺衍生物M1e,收率为69%。图6(d)展示了M1e在液相质谱ESI正模式下测试所得的谱图;由图6(d)可知,测得离子峰[M+H]+=519.3,与目标产物M1e的理论分子质量518.3一致。
M1e与对硝基氯甲酸苯酯发生酰化反应,得到稳定的三肽苯酰胺活性酯衍生物M1,收率84%,该路线总产率为27%。图6(e)展示了M1在液相质谱ESI正模式下测试所得的谱图;由图6(e)可知,测得离子峰[M+H]+=628.3,因结构中包含Boc,由此推断所测样品中化合物分子量为628.3+56,与目标产物M1的理论分子质量683.3一致,确定为目标化合物。
2.2 P1和P2合成过程分析
将含有二级胺官能团的HSP90抑制剂分别与含有活性酯的三肽连接子缩合,得到两个三肽前药P1和P2。反应路线简短,条件温和,产品收率较高,为三肽类药物的合成提供了新的方法。
2.2.1 P1液相质谱分析
采用液相质谱(LCMS)对P1进行质谱分析,分析结果如图7(a)所示,测得离子峰[M+H]+=963.5,因结构中包含Boc,由此推断所测样品中化合物分子量为963.5+43,与所设计目标产物P1的相对分子质量1005.5相吻合,确定为目标化合物。
2.2.2 P1核磁氢谱分析
采用核磁氢谱(1H NMR)对P1进行氢谱分析见图7(b),P1分子式为C54H71N9O10,图中氢的个数为71,1、2、3位置的3个氢为3个羟基峰,4、5位置的6个氢为异丙基的两个甲基的氢,6、7、8、9位置的12个氢为另外4个甲基的氢,因此可以确定为目标化合物
2.2.3 P2液相质谱分析
采用液相质谱(LCMS)对P2进行质谱分析,分析结果如图8(a)所示。测得离子峰[M+H]+=954.5,与所设计的目标产物P2的相对分子质量953.5相吻合,确定为目标化合物。
2.2.4 P2核磁氢谱分析
采用核磁氢谱(1H NMR)对P2进行氢谱分析,结果见图8(b)。P2分子式为C50H67N9O10,图中氢的个数为50,1、2、3位置的3个氢为3个羟基峰,4、5、6、7、8、9位置的18个氢为6个甲基的氢,由此可以确定为目标化合物。
2.2.5 P1和P2核磁碳谱分析
核磁碳谱(13C NMR)对P1和P2进行碳谱分析见图9,根据碳的位置和数量可以确定为目标化合物。
3 结 论
本文先通过多步反应得到HSP90i-1和HSP90i-2两种HSP90抑制剂,随后分别与三肽连接子M1进行缩合反应,得到三肽前药分子P1和P2。合成三肽连接子M1的总产率为27%,通过1H NMR、LCMS、13C NMR证明成功合成了目标化合物。本文提出的整个合成路线所使用的原料廉价易得,反应都在常温下进行,条件温和可控,总产率较高。本研究对HSP90抑制剂的毒性问题进行分析,利用前药方法优化化合物的结构,从而达到降低药物毒性,提高药物安全窗的目的,对抗肿瘤药物的发展有着重要的实践意义。
参考文献:
[1]Li Y Y, Zhang T, Schwartz S J, et al. New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential[J]. Drug Resistance Updates, 2009, 12(1/2): 17-27.
[2]Sidera K, Patsavoudi E. HSP90 inhibitors: Current development and potential in cancer therapy[J]. Recent Patents on Anti-Cancer Drug Discovery, 2013, 9(1): 1-20.
[3]Alarcon S V, Mollapour M, Lee M J, et al. Tumor-intrinsic and tumor-extrinsic factors impacting hsp90- targeted therapy[J]. Current Molecular Medicine, 2012, 12(9): 1125-1141.
[4]张钟元, 阎爱侠. Hsp90抑制剂的研究进展[J]. 中国医药生物技术, 2021, 16(1): 41-50.
[5]Shrestha L, Bolaender A, Patel H J, et al. Heat shock protein (HSP) drug discovery and development: Targeting heat shock proteins in disease[J]. Current Topics in Medicinal Chemistry, 2016, 16(25): 2753-2764.
[6]Liu N X, Chen Q H, Zhang Q Q, et al. The application of prodrug-based drug delivery strategy in anticancer drugs[J]. Current Topics in Medicinal Chemistry, 2021, 21(24): 2184-2204.
[7]Zawilska J B, Wojcieszak J, Olejniczak A B. Prodrugs: A challenge for the drug development[J]. Pharmacological Reports, 2013, 65(1): 1-14.
[8]Zhang T, Yang X R, Xu W P, et al. Heat shock protein 90 promotes RNA helicase DDX5 accumulation and exacerbates hepatocellular carcinoma by inhibiting autophagy[J]. Cancer Biology & Medicine, 2021, 18(3): 693-704.
[9]Daneri-Becerra C, Galigniana M D. The Hsp90-binding immunophilin FKBP52 enhances neurodifferentiation and neuroregeneration in murine models[J]. Neural Regeneration Research, 2022, 17(3): 555-556.
[10]林智才, 陈燕丽, 苏小清, 等. Hsp90抑制剂减弱肝癌耐药细胞株对索拉非尼诱导铁死亡的耐受性[J]. 现代肿瘤医学, 2022, 30(21): 3862-3868.
[11]Rautio J, Kumpulainen H, Heimbach T, et al. Prodrugs: Design and clinical applications[J]. Nature Reviews Drug Discovery, 2008, 7(3): 255-270.
[12]Diez-Torrubia A, García-Aparicio C, Cabrera S, et al. Application of the dipeptidyl peptidase IV (DPPIV/CD26) based prodrug approach to different amine-containing drugs[J]. Journal of Medicinal Chemistry, 2010, 53(2): 559-572.
[13]García-Aparicio C, Bonache M C, De Meester I, et al. Design and discovery of a novel dipeptidyl-peptidase IV (CD26)-based prodrug approach[J]. Journal of Medicinal Chemistry, 2006, 49(17): 5339-5351.
[14]Velzquez S, de Castro S, Diez-Torrubia A, et al. Dipeptidyl-peptidase IV (DPP IV/CD26)-activated prodrugs: A successful strategy for improving water solubility and oral bioavailability[J]. Current Medicinal Chemistry, 2015, 22(8): 1041-1054.
[15]Brough P A, Aherne W, Barril X, et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer[J]. Journal of Medicinal Chemistry, 2008, 51(2): 196-218.
[16]Santos C R, Capela R, Pereira C S G P, et al. Structure-activity relationships for dipeptide prodrugs of acyclovir: Implications for prodrug design[J]. European journal of medicinal chemistry, 2009, 44(6): 2339-2346.
[17]王薇, 薛從建, 陈秋缘, 等. 间苯二酚类热休克蛋白90抑制剂关键中间体5-异丙基-2,4-二甲氧基苯甲醛合成研究[J]. 福建医科大学学报, 2021, 55(5): 373-376.
(责任编辑:刘国金)
收稿日期: 2023-01-18网络出版日期:2023-06-07
作者简介: 刘文金(1995- ),男,甘肃天水人,硕士研究生,主要从事有机物合成方面的研究。
通信作者: 曹小冬,E-mail: sheldon.cao@eubulusbio.com
猜你喜欢
肿瘤防治研究(2023年2期)2023-03-08
食管疾病(2022年1期)2022-11-26
动物营养学报(2022年3期)2022-03-30
天然产物研究与开发(2019年8期)2019-09-05
医学理论与实践(2019年8期)2019-02-26
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
医学研究杂志(2015年9期)2015-07-01
食品工业科技(2015年5期)2015-03-24
云南中医学院学报(2014年2期)2014-11-07
食品工业科技(2014年22期)2014-03-07
