
Rapid formation of Csp3–Csp3 bonds through copper-catalyzed decarboxylative Csp3–H functionalization
2023-03-14 06:52WenwenCuiYuLiXufengLiJunxinLiXiuynSongJinLvYunYeJingDoshnYng
Chinese Chemical Letters 2023年1期
Wenwen Cui,Yu Li,Xufeng Li,Junxin Li,Xiuyn Song,Jin Lv,Yun-Ye Jing,Doshn Yng,d,*
a Key Laboratory of Optic-Electric Sensing and Analytical Chemistry for Life Science,MOE,College of Chemistry and Molecular Engineering,Qingdao University of Science and Technology,Qingdao 266042,China
b School of Chemistry and Chemical Engineering,Qufu Normal University,Qufu 273165,China
c Zhejiang Wansheng Co.,Ltd.,Linhai 317000,China
d Key Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology (Ministry of Education),Department of Chemistry,Tsinghua University,Beijing 100084,China
Keywords:Copper Cross-coupling Csp3-H functionaliztion Decarboxylation Csp3–Csp3 bond formation
ABSTRACT Transition-metal-catalyzed decarboxylative and C–H functionalization strategy for the construction of Csp2-Csp2,Csp2-Csp,and Csp2-Csp3 bonds has been extensively studied.However,research surveys of this synthetic strategy for the Csp3-Csp3 bond forming reactions are surprisingly scarce.Herein,we present an efficient approach for the rapid formation of Csp3–Csp3 bond through copper-catalyzed decarboxylative Csp3–H functionalization.The present method should provide a useful access to C3-substituted indole scaffolds with possible biological activities.Mechanistic experiments and DFT calculations supported a dual-Cu(II)-catalytic cycle involving rate-determining decarboxylation in an outer-sphere radical pathway and spin-crossover-promoted C–C bond formation.This strategy offers a promising synthesis method for the construction of Csp3–Csp3 bond in the field of synthetic and pharmaceutical chemistry and extends the number of still limited copper-catalyzed decarboxylative Csp3–Csp3 bond forming reaction.
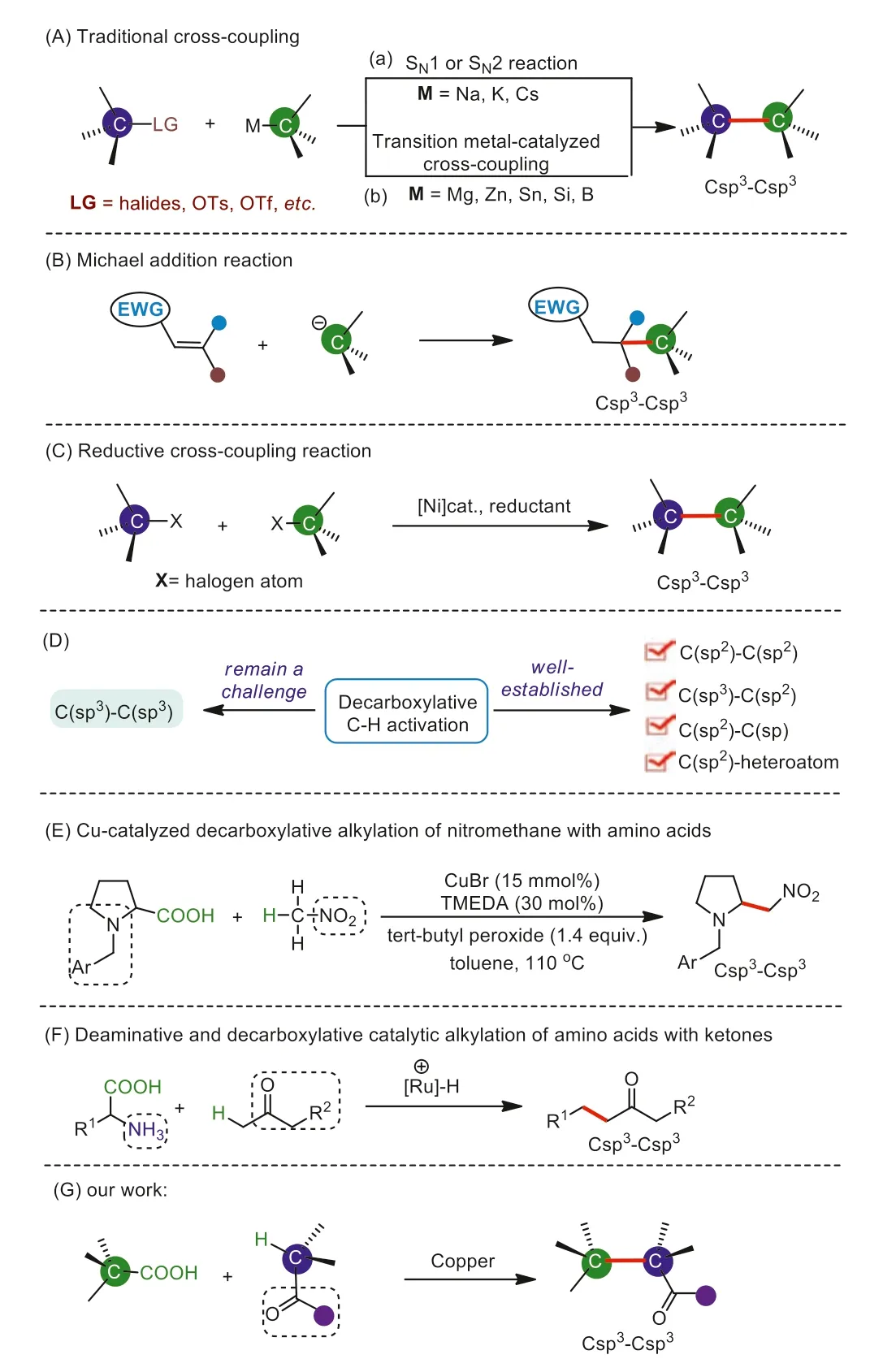
Csp3–Csp3linkages are one of the most basic chemical bonds,which are ubiquitous in a wide range of pharmaceuticals and naturally occurring products [1–3].The introduction of Csp3moiety with three-dimensional spread not only increases the lipophilicity,but also the hydrophilicity of the compound.As a consequence,a statistical correlation between the complexity of molecules and clinical success has been proven to be related to the number of Csp3moieties in a drug candidate [4].Because of its importance in drug discovery,seeking direct and selective crosscoupling approaches for the construction of Csp3-Csp3bonds has been of continuous interest in pharmaceutic and organic chemistry.Several classical methods for the construction of Csp3-Csp3bond have been developed,including: (1) nucleophilic substitution reactions between carbon anions and alkyl halides (Scheme 1Aa) [5]; (2) metal-catalyzed traditional cross-couplings or radicaltype cross-couplings between alkyl-metallic reagents such as alkylmagnesium,-zinc,or -boranes and alkyl halides (Scheme 1B) [6–12]; (3) Michael addition reaction between carbon anions and activated olefins (Scheme 1C) [13]; and (4) reductive cross-coupling between two different alkyl halides [14–16].Despite great achievements have been made in this field,these developed methods generally suffer from unavailable of starting substrates,harsh reaction conditions,and narrow substrate scopes.Therefore,significant space and challenges still exist for the construction of Csp3-Csp3bond with regard to generality,reaction conditions,and catalytic efficiency.
Carboxylic acids are common chemical raw materials and widely found in natural products,and biologically active molecules[17].Recently,decarboxylative couplings for the formation of C–C and C-heteroatom bonds has been made great achievements owing to that carboxylic acids are easily prepared,easy to store,stable to air and moisture,and besides that,CO2is the sole waste product from the decarboxylative transformation [18–38].On the other hand,direct activation/functionalization of C–H bonds has emerged as an environmentally friendly and economical alternative to traditional coupling reactions.Over the past few decades,great efforts have been made to develop new strategies for the direct C–H functionalizations [39–46].The combination of decarboxylation and C–H functionalization strategy for the construction of C–C bond,which possesses the common advantages of both,has been demonstrated as a powerful and convenient synthetic tool in organic synthesis.In this respect,most of these reactions are mainly focused on the construction of Csp2-Csp2bond,Csp2-Csp bond,and Csp2-Csp3bond [47–53].However,research surveys of this synthetic strategy for the Csp3-Csp3bond forming reactions are surprisingly scarce.This is probably due to the following reasons: (1) The R-[M] species formed from aliphatic carboxylic acids is usually not stable enough,thus is prone to undergo self-coupling reaction or other side reactions; (2) Csp3-H bonds are less polar,thus have weaker coordination to metal catalysts,making them difficult to be activated; (3) Csp3-H bonds widely exist in one molecule,making the selective cleavage difficult.However,there are still sporadic reports of Csp3-Csp3bond construction based on decarboxylative and C–H functionalization strategy.In 2009,Li and Liang reported the first CuBr-catalyzed decarboxylative Csp3-Csp3bond coupling reaction usingα-amino acids as starting materials[54].In 2013,Yi and co-workers developed a novel and efficient Ru-catalyzed alkylation method using readily available amino acid substrates as a bio-based alkylation reagent [55].Therefore,there is still plenty of room to develop more efficient decarboxylative and C–H functionalization strategies for the construction of Csp3-Csp3bond using aliphatic carboxylic acids as alkylating reagents.
Scheme 1.Strategies for Csp3-Csp3 bond formation.
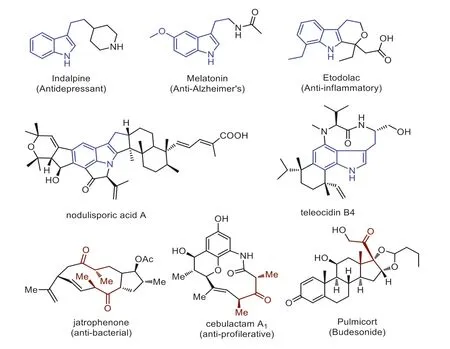
Scheme 2.C3-substituted indole and α,α-di-substituted ketone skeletons in natural products and biological molecules.
Indole is a privileged fragment,serving as an important building block in the construction of pharmaceuticals,natural products,and functional materials [56].Especially,the C3-substituted indole derivatives widely occur in natural products and biologically active molecules,such as antidepressant,anti-Alzheimer,and antiinflammatory (Scheme 2) [57–60].In addition,branchedα,α-disubstituted ketones are also an important class of bioactive functional units and synthetic building blocks in multi-step organic synthesis (Scheme 2) [61–63].We envisaged that combining the frameworks of C3-substituted indoles andα,α-di-substituted ketones might yield valuable substrates for the synthesis of biologically active compounds with different structural features from the two units separately.Inspired and encouraged by these excellent works of decarboxylative and C–H functionalization,we herein report a novel and efficient approach for the rapid construction of Csp3–Csp3bonds through copper-catalyzed decarboxylative Csp3-H functionalization strategy between ketones and 3-indoleacetic acids (Scheme 1G).
We commenced our study by examining the reaction between deoxybenzoin (1a) and 2-(1H-indol-3-yl)acetic acid (2a) to investigate reaction conditions including the optimization of catalysts,ligands,oxidants,solvents,bases,and temperature under a nitrogen atmosphere.As shown in Table 1,six copper catalysts (entries 1–6) were tested at 120°C in the presence of 0.2 equiv.of 4,4′-di–tert–butyl–2,2′-bipyridine (dtbpy) (A) as the ligand (relative to the amount of 1a) in DMSO,and Cu(OAc)2exhibited the highest reaction activity (entry 1).The reaction could not take place in the absence of a catalyst (entry 7).Next,different solvents including DMF,and NMP were examined,and DMF was found to be the best choice (entry 1vs.entries 8 and 9).In addition,we compared various oxidants such as KMnO4,AgOAc,and Ag2O,and MnO2was showed the best result.Only 29% yield of the target product 3a was obtained in the absence of a oxidant (entry 13).Furthermore,various ligands were screened (entries 1,13–19),and 2,2′:6′,2′′-terpyridine (TPY) (D) exhibited the highest efficiency (entry 17).In order to increase the yield of the reaction,various bases were investigated,and K3PO4was superior to the others (entries 21–24).Finally,the effect of temperature was also investigated (entries 19–21),and the yields reached the maximum when the temperature was 90 °C.
With the optimal reaction conditions in hand,we began to investigate the scope and generality of the copper-catalyzed decarboxylative/Csp3–H functionalization reaction between deoxybenzoins 1 and 3-indoleacetic acids 2,and the results are summarized in Scheme 3.We were pleased to find that diverse deoxybenzoins 1 bearing either electron-donating groups,or electronwithdrawing groups smoothly reacted with 3-indoleacetic acids 2,affording the corresponding alkylation products in good to excellent yields.The hindrance effect of this decarboxylative/Csp3–H functionalization transformation was not obvious; the deoxybenzoins bearing methyl at different positions could react with 3-indoleacetic acids efficiently (3e and 3f).Notably,a strong electron withdrawing group such as nitro was also tolerated under the reaction standard conditions (3o).It should be noted that the electroneffect of the substituted groups in 3-indoleacetic acids including electron-rich,-deficient,and -neutral groups did not display evidently difference of reactivity.
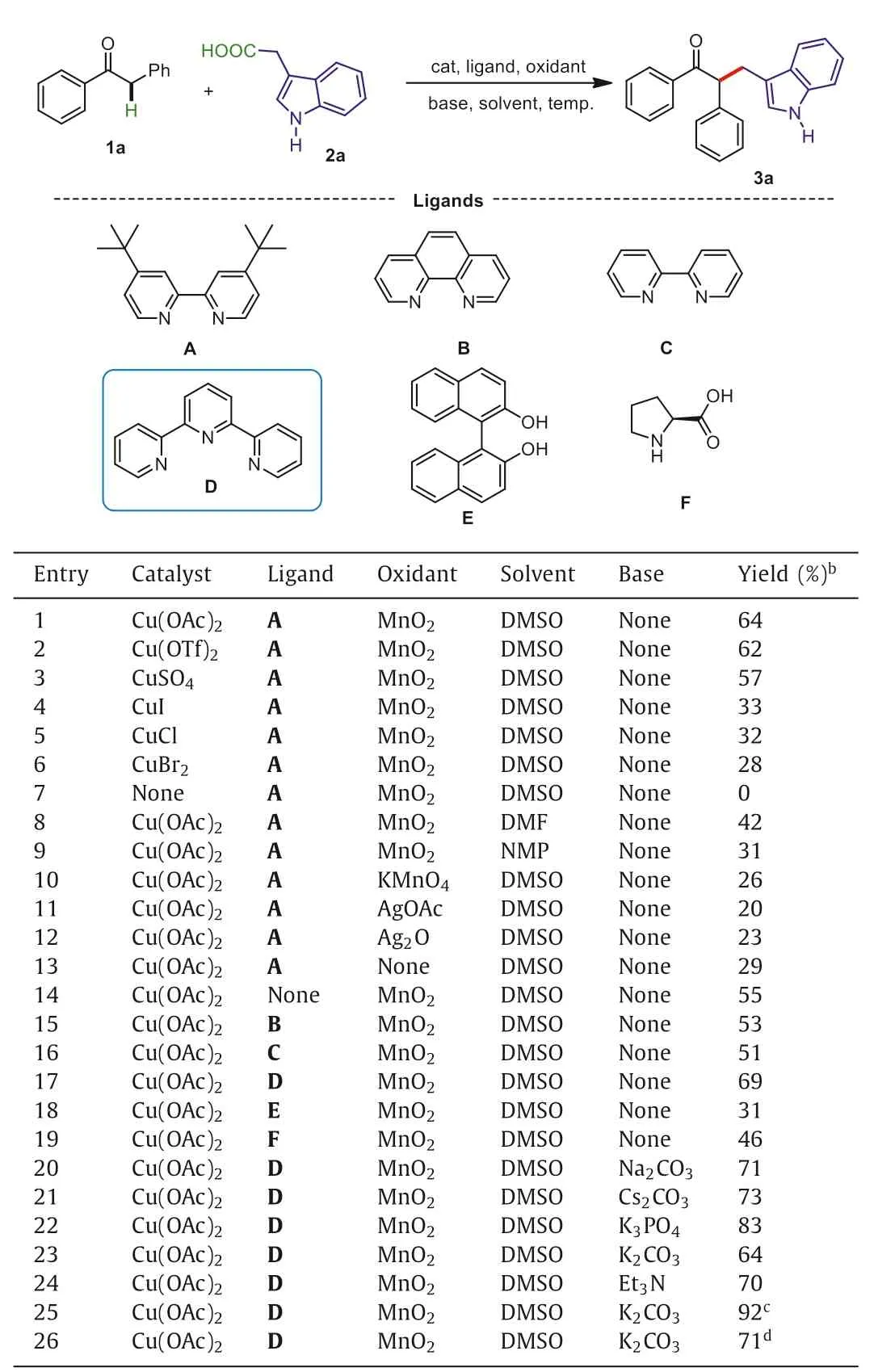
Table 1 Optimization of the reaction conditions.a,b
Subsequently,the coupling reactions of other ketones such as 1,3-diphenylpropan-1-ones and 1-phenylpropan-2-ones with 3-indoleacetic acids 2 were evaluated in the present transformation(Scheme 4).To our delight,the reaction proceeded well,and afforded the desired products in moderate yield (5a–5h).In addition,1,3-dicarbonyl compounds are also amenable to the reaction,delivering the products (5i–5l) in good yields.Furthermore,the present transformation could tolerate some functional groups such as methoxyl groups,methyl groups,NO2group,CF3group,and CBr bond,which provided great opportunities for further modifications.
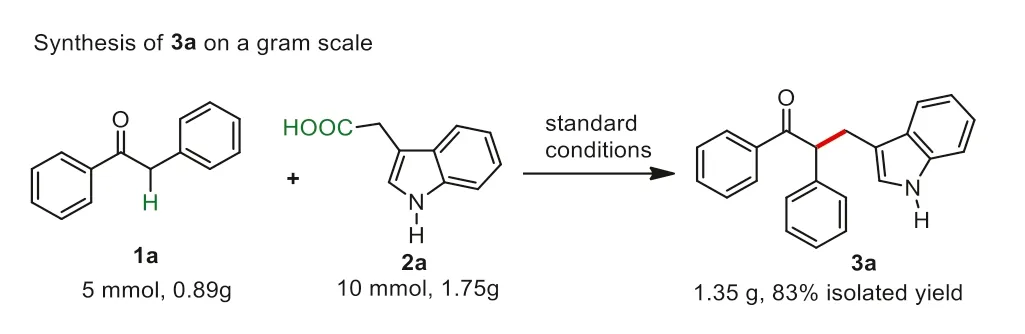
Next,gram scale applications for the copper-catalyzed decarboxylative/Csp3–H functionalization reaction between deoxybenzoin 1a and 3-indoleacetic acid 2a were investigated.As shown in Scheme 5,the proposed reaction proceeded smoothly under the standard conditions,which could afford 1.35 g of 3a in 83% yield.Therefore,this copper-catalyzed protocol could be used as a practical approach for the synthesis of alkylating ketones.
Several control experiments were conducted to investigate the mechanism.First,in order to demonstrate the role of the Cu(II)-based catalyst,the copper complex 9 was synthesized according to the previous report [64].This copper catalyst 9 was then applied in place of the combination of Cu(OAc)2and TPY as the catalyst for the reaction.To our delight,the desired product 3a was obtained in almost the same yield as that under the standard conditions.This preliminary experimental result indicated that this Cu(II) complex generated in situ was the active catalyst in the present transformation (Scheme 6A).When 2 equiv.of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy,a well-known radical capture) was added to the reaction system,the reaction was completely suppressed and a TEMPO-trapped complex 9 was detected by HRMS analysis (see Supporting information for details),thus indicating that a radical process might be involved in the present transformation (Scheme 6B).When the reaction of 1a with 2a was carried out in the absence of MnO2,the present reaction proceeded sluggishly and gave the desired product 3a in 24% isolated yield only,suggesting that MnO2might be used as an oxidant for the copper catalyst (Scheme 6C).Treatment of 1a with 2-(1H-indol-3-yl)acetaldehyde 2aa under the standard conditions did not afford the product 3a,indicating 2aa was not the intermediate in the present transformation (Scheme 6D).
Furthermore,to gain mechanistic insights into this coppercatalyzed Csp3–Csp3bond forming reaction,a kinetic isotope effect (KIE) study was performed.As shown in Scheme 7,two parallel reactions of deoxybenzoin 1a and D-1a with 3-indoleacetic acid 2a were carried out,and no kinetic isotope effect (kH/kD=1.81017/1.01695=1.78) was observed,which suggested that Csp3–H bond cleavage might not take place during the turnover-limiting step [65].
The mechanistic details were further clarified with the aid of computational methods (see Supporting information for more details).The potassium acetate 2a’ can bein situgenerated from the reaction of 2a and K3PO4with a free energy change of-19.1 kcal/mol (Scheme S1 in Supporting information),and then undergoes anion exchange with Int1 to form copper(II) acetate Int2 with an energy decrease of 5.0 kcal/mol (Fig.1) [53,66,67].In contrast to the inner-sphere anionic mechanism [68],Int2 undergoes decarboxylationviaan outer-sphere radical mechanism (TS1)[69,70],in which no Cu–C bond forms.The decarboxylation has a free energy barrier of 28.7 kcal/mol and generates CO2,radical R1 and Int3.The radical rebound of R1viaTS2 to generate Int4 is fast but is slightly exergonic by only 2.1 kcal/mol,meaning that this step can be reversible at the reaction temperature.This result is consistent with the free radical capture experiment (Scheme 6B).In the next,the Cα–H bond of 1a is activated by another Int1viaTS3 to generate copper(II) enolate Int6 with an energy barrier of 23.2 kcal/mol.Then R1 is released again from Int4viaTS2,and combines with Int6 to form the triplet complex Int7-T.From Int7-T,the outer-sphere C–C bond formationviathe triplet transition state TS4-T is less likely as the corresponding overall energy barrier is 32.1 kcal/mol.It was found that the geometry optimization starting from Int7-T as singlet minimum spontaneously forms the C–C bond to afford Int8.Inspired by this phenomenon and our previous works [71,72],a spin-crossover pathwayviathe minimum energy crossing point MECP1 that turns the triplet Int7-T into singlet Int8 was proposed for the C–C bond formation (Geometry optimization starting from MECP1 as singlet minimum also spontaneously formed Int8).The electronic energy of MECP1 is higher than that of Int7-T by only 5.2 kcal/mol,indicating that this step is feasible.We also considered C–C reductive elimination from a Cu(III) complex but this pathway seems to be less favored according to the estimated energy barrier (about 32.6 kcal/mol,Fig.S4 in Supporting information).The calculated energy profile indicates that the decarboxylation is the rate-determining step,and is in line with the absence of H/D primary kinetic isotope effect(Scheme 7).
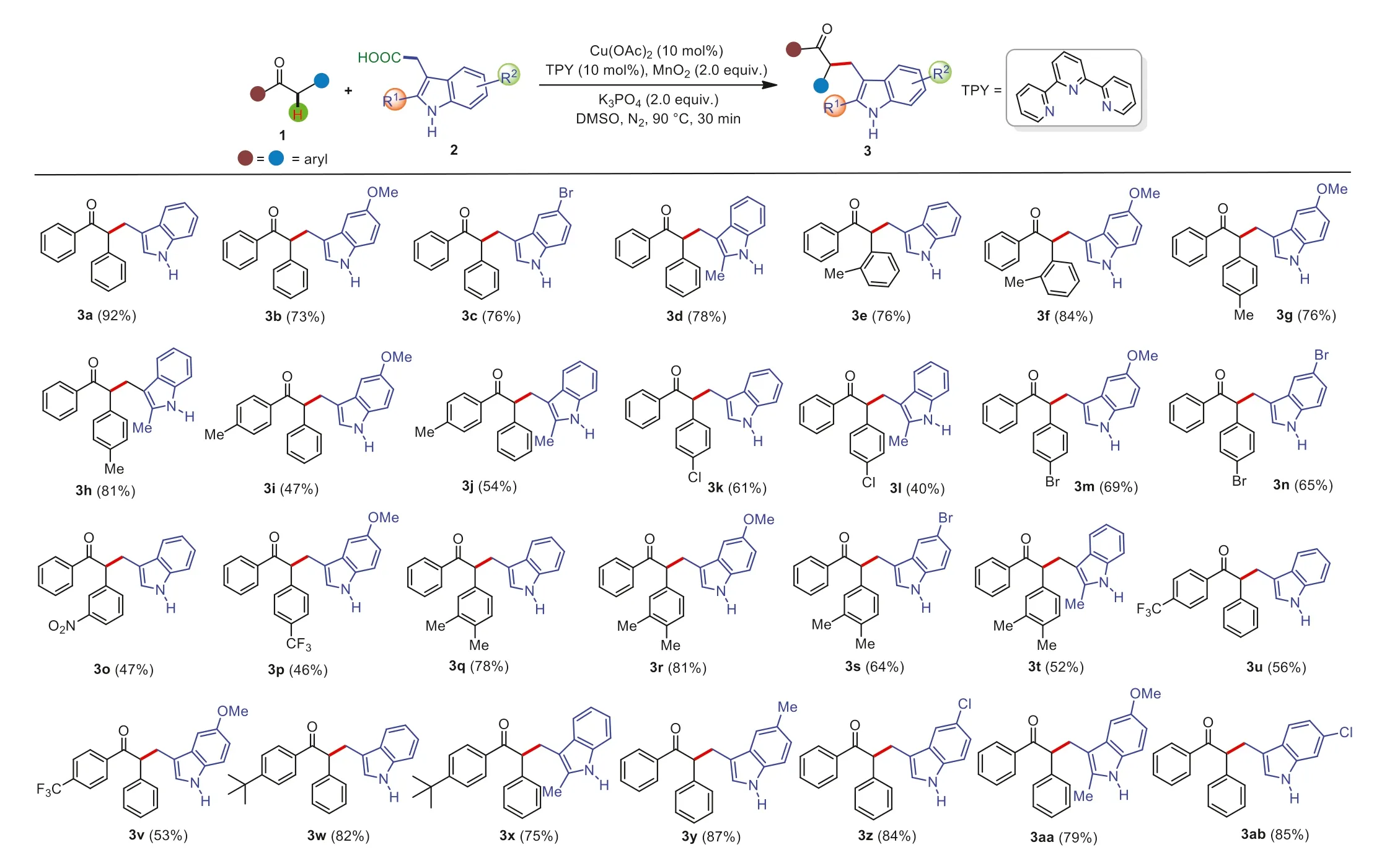
Scheme 3.Substrate scope of the coupling of deoxybenzoins with 3-indoleacetic acids.Reaction conditions: deoxybenzoins 1 (0.2 mmol),3-indoleacetic acids 2 (0.4 mmol),Cu(OAc)2 (0.02 mmol),2,2′:6′,2′′-terpyridine (TPY) (0.02 mmol),MnO2 (0.4 mmol),K3PO4 (0.4 mmol),DMSO (2.0 mL),90 °C,reaction time (0.5 h).Isolated yield.
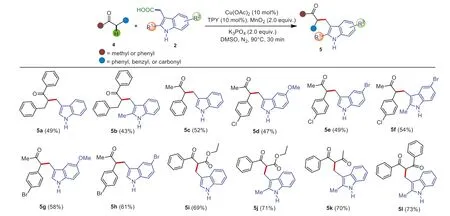
Scheme 4.Substrate scope of the cross-coupling of other ketones with 3-indoleacetic acid.Reaction conditions: 2-Phenylacetophenones 1 (0.2 mmol),3-indoleacetic acids 2(0.4 mmol),Cu(OAc)2 (0.02 mmol),TPY (0.02 mmol),MnO2 (0.4 mmol),K3PO4 (0.4 mmol),DMSO (2.0 mL),90 °C,reaction time (0.5 h).Isolated yield.
Scheme 5.Synthetic applications.
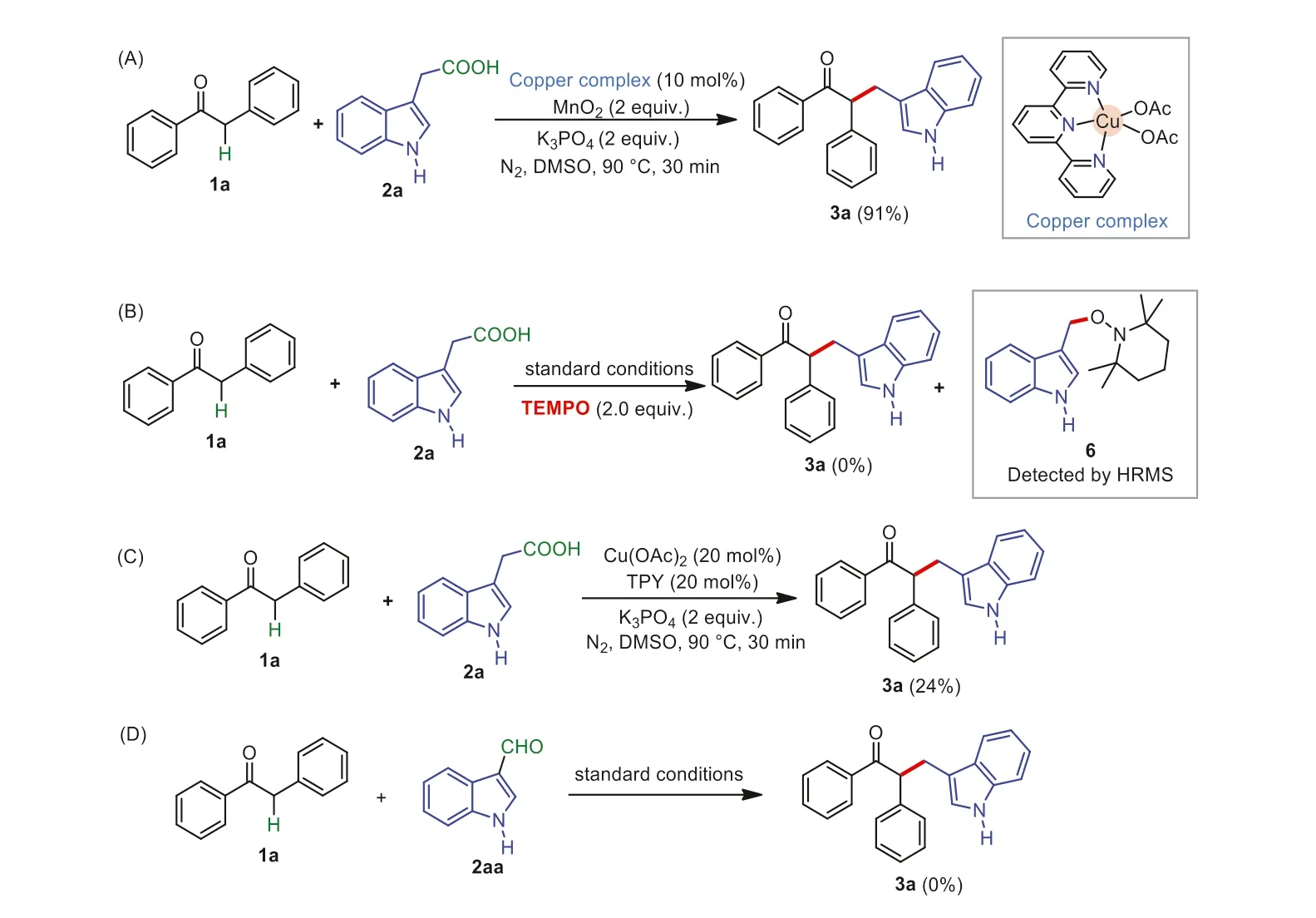
Scheme 6.Control experiments.
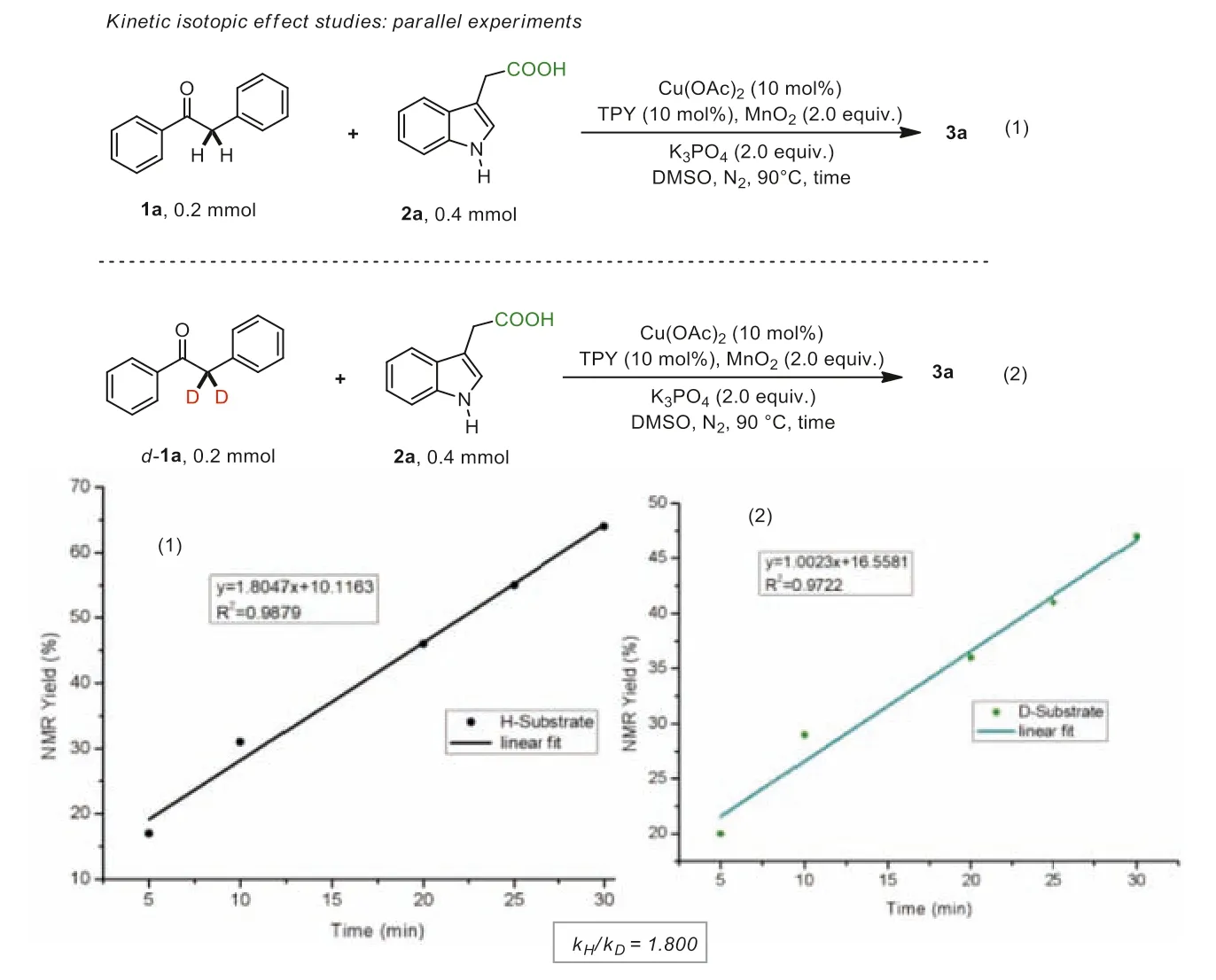
Scheme 7.Mechanistic studies.
Based on these experimental findings,a possible reaction mechanism for this copper-catalyzed decarboxylative cross-coupling pathway was proposed in Scheme 8.First,treatment of Cu(OAc)2with TPY produces a chelated Cu(II) complex LCu(II).Next,ligand exchange reaction of LCu(II) with 2 leads to the intermediate A.Then,the intermediate A is decarboxylated to deliver the active copper species B,which undergoes homolytic reaction to give the radical C and LCu(I).Meanwhile,the substrate 1a reacts with LCu(II) with the assistance of a base to afford the intermediate Cu(II)-O-enolate D.Subsequently,the intermediate D reacts with radical C to give the desired product 3,together with a release of the LCu(I) complex.Finally,the LCu(I) complex is oxidized to a catalytic LCu(II) species by MnO2to finish the catalytic cycle.
In summary,we have established a new strategy for constructing the Csp3–Csp3bonds through copper-catalyzed decarboxylative Csp3–H functionalization.A series of potentially biological C3-substituted indole scaffolds could be efficiently and conveniently obtained in moderate and good yields with excellent functional group tolerance.Preliminary mechanistic experiments and DFT calculations suggest that this reaction was likely to proceedviaCu(II)-catalyzed rate-determining outersphere radical decarboxylation while the C–C bond formation is achieved through a spin-crossover pathway.We anticipate that this strategy will open a new avenue for the formation of Csp3–Csp3bonds and will also find wide applicability on synthetic and pharmaceutical chemistry.Further investigations on the practical application of this method wre ongoing in our laboratory.
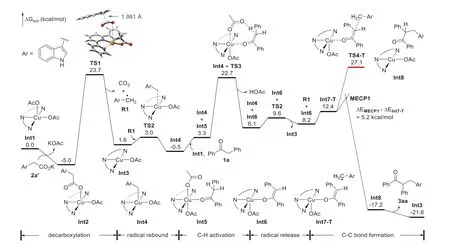
Fig.1.Calculated relative solution-phase Gibbs free energies of Cu-catalyzed decarboxylative Csp3–Csp3 cross-coupling of deoxybenzoin and 3-indoleacetic acid (kcal/mol).
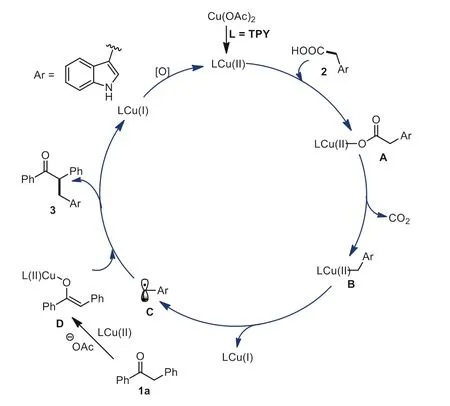
Scheme 8.Possible reaction pathway.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No.21702119),Natural Science Foundation of Shandong Province (Nos.ZR2016JL012,ZR2020JQ07),and the Scientific Research Foundation of Qingdao University of Science and Technology (No.1203043003457).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.04.075.
 Chinese Chemical Letters2023年1期
Chinese Chemical Letters2023年1期
- Chinese Chemical Letters的其它文章
- Diabetic wound healing activated by supramolecular cascade reaction
- MBenes: Two-dimensional transition-metal borides with ordered metal vacancies
- Wet-adhesive materials of oral and maxillofacial region: From design to application
- Diverse catalytic systems for nitrogen-heterocycle formation from O-acyl ketoximes
- Fluorine-containing drugs approved by the FDA in 2021
- The development and application of dual-comb spectroscopy in analytical chemistry