
Fluorine-containing drugs approved by the FDA in 2021
2023-03-14 06:51JingruiZiyiLiGgnDhwnWiZhngAlxnrSorochinskyGrgButlrVimSoloshonokJinlinHn
Chinese Chemical Letters 2023年1期
Jingrui H,Ziyi Li,Ggn Dhwn,Wi Zhng,Alxnr E.Sorochinsky,Grg Butlr,Vim A.Soloshonok,Jinlin Hn,*
a Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources,International Innovation Center for Forest Chemicals and Materials,College of Chemical Engineering,Nanjing Forestry University,Nanjing 210037,China
b Department of Biomedical Science,Acharya Narendra Dev College,University of Delhi,New Delhi 110019,India
c Department of Chemistry,University of Massachusetts Boston,Boston,MA 02125,United States
d V.P.Kukhar Institute of Bioorganic Chemistry and Petrochemistry,The National Academy of Sciences of Ukraine,Kyiv 02094,Ukraine
e Oakwood Chemical,Inc.,Estill,SC 29918,United States
f Department of Organic Chemistry I,Faculty of Chemistry,University of the Basque Country UPV/EHU,20018 San Sebastián,Spain
g IKERBASQUE,Basque Foundation for Science,Plaza Bizkaia,48013 Bilbao,Spain
Keywords:Fluorine-containing compounds Blockbuster drugs Pharmaceuticals Anti-cancer Drug design and development Asymmetric synthesis
ABSTRACT Nine new fluorine-containing drugs have been approved by the US Food and Drug Administration (FDA)in 2021,which are presented in this review article.These small molecular drugs feature aromatic fluorine,trifluoromethyl and chlorodifluoro groups.The therapeutic areas of these fluorine-containing drugs include multiple myeloma,lymphoma,HIV,chronic heart failure,chronic myeloid leukemia,(ANCA)-associated vasculitis,migraines,von Hippel-Lindau disease,and non-small cell lung cancer.The brief biological activities and the synthetic methods have been discussed in this review for each of these nine drugs.
1.Introduction
For over 20 years,organo-fluorine molecules represent one of the most fast-growing classes of organic compounds [1–17].The role of fluorine in the design of pharmaceutical drugs [18–31],agrochemicals,and specialty materials [32–36] is well-recognized.Historically,efforts have been made to incorporate Fluorine into biologically active compounds to develop new pharmaceutical drugs and formulations [18–31].Scientists pay special attention to the records pertinent to new pharmaceutical drugs and the aspects of their design and therapeutic activity.The goal of this review article is to profile nine new fluorine-containing drugs 1–9 introduced to the market in 2021,which include melphalan flufenamide(PepaxtoTM),umbralisib (UkoniqTM),cabotegravir and rilpivirine(CabenuvaTM),vericiguat (VerquvoTM),asciminib (ScemblixTM),atogepant (QuliptaTM),avacopan (TavneosTM),belzutifan (WeliregTM)and sotorasib (LumakrasTM) (Fig.1).Where it is possible,the mode of biological activity is discussed,emphasizing the specific role of fluorine in the development of a particular drug.For each compound,synthetic routes will be discussed as well as the manner of fluorine introduction into the molecule.
2.Melphalan flufenamide (Melflufen,pepaxtoTM)
Melflufen 1 is an ester of an alkylated dipeptide,which was developed by Oncopeptides for the treatment of myeloma and amyloid light chain amyloidosis [37].Melflufen belongs to a new class of peptidase enhancing compounds,which targets the process of multiple myeloma tumor transformation with a unique mechanism.With the use of a simple peptide bond,melflufen’s activity is realized by aminopeptidase,resulting in peptidase enhancement effect [38].Melflufen showed antitumor activity in multiple myeloma,lymphoma and acute myeloid leukemia cell lines,and primary tumor cells [37,39].Melphalan flufenamide contains a key part of melphalan 10 and apara-fluoro-L-phenylalanine (Fig.2).Structure-activity relationship (SAR) studies disclosed that melphalan flufenamide showed more than ten-fold pharmacological activity compared to melphalan in the cell lines of RPMI 8226,CCRF-CEM and NCI-H69.Although the analogs 11–13 featuring other phenylalanine substitutions (hydroxyl,amino and methoxyl)showed very similar activity with that of melphalan flufenamide 1,the introduction of fluorine substitution increases the metabolic stability [40].The results of the phase 2 HORIZON study support the US Food and Drug Administration (FDA) to accelerate the approval of melflufen in 2021 for the treatment of three types of refractory multiple myeloma patients [41].
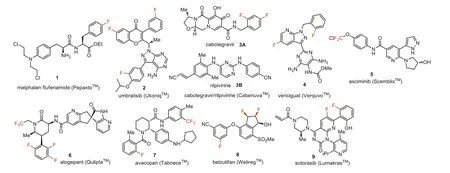
Fig.1.Structures of approved fluorine-containing drugs.
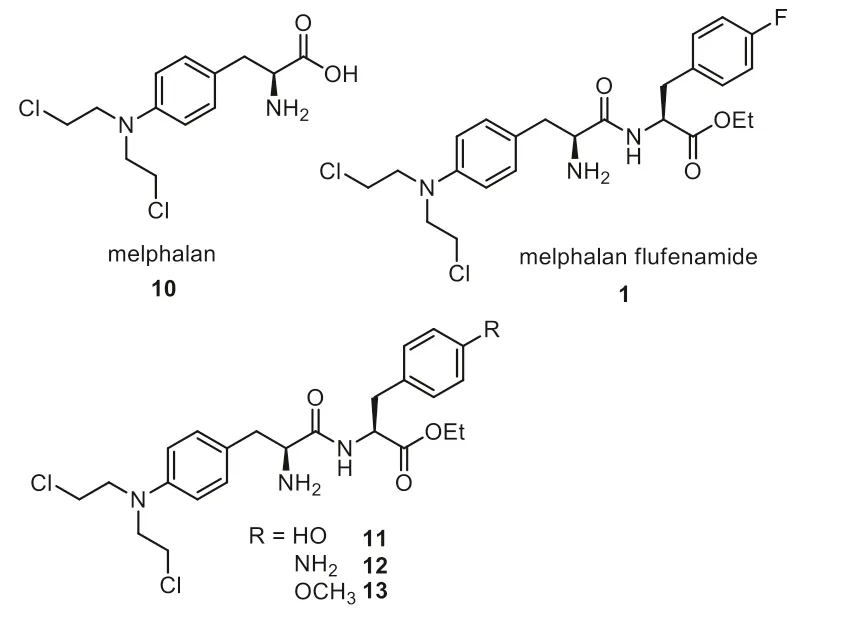
Fig.2.Structures of melflufen analogs.
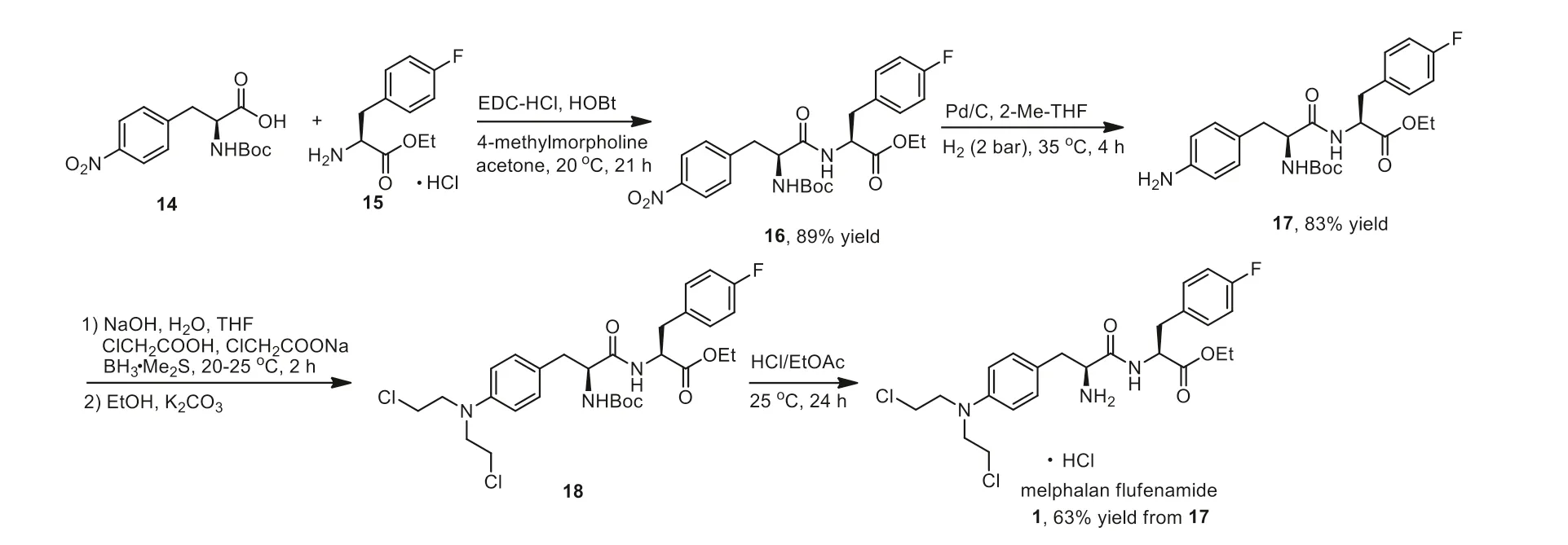
The original chemistry for the synthesis of melphalan flufenamide 1 used melphalan and 4-fluoro-L-phenylalanine ethyl ester as the starting materials.The synthetic strategy was in milligram scale and gave a poor yield of the hydrochloride salt melflufen.However,this method provided enough material for SAR studies [42].In 2019,Magle Chemoswed developed a safe process for manufacturing of melflufen 1 on a kilogram scale,which was shown in Scheme 1.The condensation reaction between L-Boc-4-nitrophenylalanine (14) and L-4-fluoro-phenylalanine ethyl ester hydrochloride salt (15) employed 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC–HCl) and 1-hydroxybenzotriazole (HOBt) as condensation reagents in the presence ofN-methylmorpholine afforded the amide 16 in 89% yield after 21 h.The amide 16 was reduced using the catalyst Pd/C under hydrogen atmosphere at 35 °C to give the free amine 17 in 83% yield.Reductive alkylation of amine 17 with the use of chloroacetic acid and sodium chloroacetate as the alkylating reagents and borane-dimethyl sulfide as the reductant afforded the intermediate 18.Treatment with potassium carbonate in ethanol rendered the alkylated intermediate 18 which then underwent deprotection in the presence of HCl in ethyl acetate to generate the target melflufen chloric acid salt 1 at room temperature after 24 h.Notably,alkylated pharmacophores do not involve toxic substances in the whole synthesis process,which makes this method more efficient and safer [43].
3.Umbralisib (UkoniqTM)
Umbralisib 2 a tri-fluorinated chiral compound,was developed by Rhizen and TG Therapeutics as a next-generation of dual phosphoinoside-3-kinaseδinhibitor (PI3Kδ) and casein kinase 1 epsilon inhibitor (CK1ε) [44].In particular,there are still no known clinically relevant drug-drug interactions observed for umbralisib[45].Umbralisib has a unique chemical structure (Fig.3),which contains a 4H-chromen-4-one,a 1H-pyrazolo[5,4-d]pyrimidine and fluorinated phenyl rings.Preclinical analysis showed that umbralisib can effectively inhibit PI3Kδat a clinically achievable concentration compared with other approved phosphoinoside-3-kinase inhibitors [46].Umbralisib exhibits great selectivity for the PI3Kδisoform with more than 1500-fold overα- andβ-isoforms(>10,000 nmol/L and>10,000 nmol/L,respectively,the dissociation constant [Kd]=6.2 nmol/L) [45].Umbralisib is unique in the inhibiting of casein kinase 1 epsilon,which plays a key role in the protein translation of oncogenes and may affect the immune regulation of T cells [46].
Rhizen also conducted SAR studies [47–49].The results disclose that the fluorine substitution on phenyl ring and the absolute configuration play key roles for the inhibitory activity of PI3Kδand selectivity over PI3K isoforms.For example,IC50(PI3Kδ) value for analog 19 with non-fluoro substitution on 4H-chromen-4-one was 13.83 nmol/L,with>1000 fold selectivity over PI3Kα.While IC50(PI3Kδ) value for umbralisib 7 is 22.23 nmol/L with more than 10,000 fold over PI3Kα.The (R)-isomer 20 showed a decreased activity with an IC50value of 1447 nmol/L [49].Umbralisib received its first approval from FDA in February 2021 with the trade name as Ukoniq,which is specifically used for the treatment of adult patients with relapsed or refractory marginal zone lymphoma (MZL)[44–50].
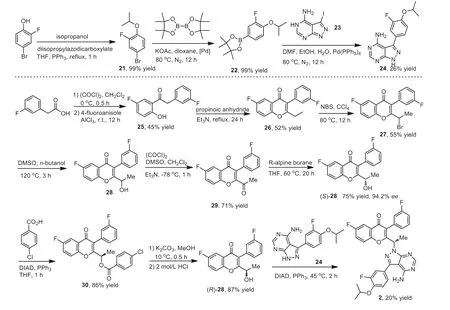
Rhizen and TG Therapeutics patented a synthetic method for the preparation of umbralisib 2 with 1H-pyrazolo[5,4-d]pyrimidine 24 and chiral alcohol 28 as the key intermediates (Scheme 2).The reaction of 4–bromo-2-fluorophenol and isopropanol in the presence of diisopropyl azodicarboxylate (DIAD) and triphenylphosphine produced the ether 21 in 99% yield,which was subjected to a coupling reaction with bis(pinacolato)diboron with[1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium (II) as a catalyst resulting in the formation of intermediate 22.Then,Suzuki coupling reaction between intermediate 22 and 3-iodo-5Hpyrazolo[3,4-d]pyrimidin-4-amine (23) provided the key intermediate 24.
Scheme 1.Synthesis of melphalan flufenamide 1.
Scheme 2.Synthesis of umbralisib 2.
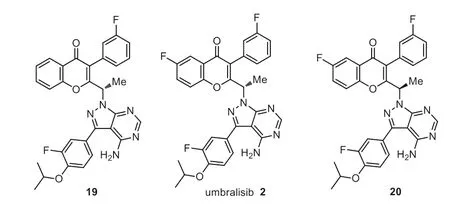
Fig.3.Structures of umbralisib and analogs.
Friedel-Crafts acylation between 4-fluoroanisole and in situ generated acyl chloride from 3-fluorophenylacetic acid afforded the intermediate 25,which underwent cyclization reaction with propionic anhydride to give the 4H-chromen-4-one intermediate 26 in 52% yield.Treatment of intermediate 26 byN-bromosuccinimide(NBS) at 0 °C for 12 h generated intermediate 27,which then was converted into alcohol 28 in the presence of DMSO andn-butanol.Swern oxidation followed by asymmetric reduction by R-alpine borane afforded the (S)-alcohol 28 in 94.2%ee,which was converted into (R)-alcohol 28viatwo-step conversion.Finally,DIADpromoted condensation reaction between the two intermediates 24 and (R)-28,completed the synthesis of umbralisib 2 was obtained in 20% yield [48].
4.Cabotegravir and rilpivirine (CabenuvaTM)
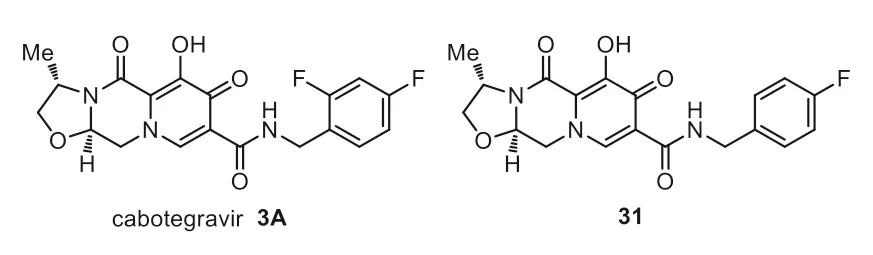
Fig.4.Structures of cabotegravir and analog.
Cabenuva (cabotegravir and rilpivirine,injectable formulation)is a long-acting (extended release) regimen,which is a combination of the integrase strand transfer inhibitor (INSTI) cabotegravir,developed by ViiV Healthcare,and the non-nucleoside reverse transcriptase inhibitor (NNRTI) rilpivirine,developed by Janssen Sciences Ireland UC.It received its first approval in March 2020 in Canada [51,52],and was approved by the FDA in January 2021 for the treatment of HIV infection.
Cabotegravir 3A is a new long-acting parenteral and a highly effective integrase inhibitor with a half-life of 54 days,allowing parenteral administration every other month.Cabotegravir features low water solubility,high activity,long half-life and slow metabolic clearance [53].Another advantage of cabotegravir relies on its main metabolization by uridine diphosphate glucuronosyltransferase 1A1,leading to less likely interactions with other antiretroviral drugs [54].Cabotegravir is a chiral compound which has a flexible six-membered ring,a rigid five-membered ring as its metal chelating scaffold,and a fluorinated benzene ring (Fig.4) [55].The SAR studies clearly showed the importance of the incorporation of fluorine substitution into cabotegravir [56].The introduction of 2-fluoro substituent brought an improved potency by approximately 4-fold compared with its analog 31 (pHIVIC50value=0.5 nmol/L and 2.1 nmol/L for 3A and 31 respectively [56].
The synthesis of cabotegravir 3A was presented in Scheme 3[57],starting from the readily available chiral alaninol as the key reagent for the introduction of chiral moiety in cabotegravir.As shown in Scheme 3,the pyridone 36 was a key intermediate for this strategy.First,β-keto ester 32 was used as the starting material,which was converted into vinyl amine 33 under reflux inN,N-dimethylformamide (DMF) and dimethylacetamide (DMA).Next the reaction of amine 33 and aminoacetaldehyde dimethyl acetal provided the vinylogous amine 34,which underwent cyclization reaction with dimethyl oxalate in the presence of LiOMe which was then hydrolized under basic conditions to yield the key pyridinone intermediate 36.Intermediate 36 can be successfully transformed into the corresponding aldehydeviathe treatment of 36 with excess HOAc and catalytic CH3SO3H,which reacted with alaninol to construct the oxazolidine ring featuring two chiral carbon centers 37 in 74% yield and 34:1 diastereoselectivity.
Intermediate 37 coupled with 2,4-difluorobenzylamine in the presence ofN,N’-carbonyldiimidazole (CDI) gave the amide 38.Treatment of 38 with various magnesium salts in acetonitrile led to completely selective C-6 demethylation to afford the desired cabotegravir 3A in 93% yield.This synthesis is highlighted by the efficient construction of highly functionalized pyridinone nuclei and the highly diastereoselective formation of acyl oxazolidine moieties [57,58].
Another component of cabenuva is rilpivirine 3B,which is a diaryl pyrimidine non-nucleoside reverse transcriptase inhibitor that inhibits HIV-1 reverse transcriptase through non-competitive binding.In the United States,rilpivirine is recommended in combination with other antiretroviral drugs for the treatment of HIV-1 infected adult patients.Rilpivirine was previously approved by FDA in 2011 with the drug name Edurant [59,60],approved with emtricitabine/tenofovir disoproxil fumarate with the drug name Complera in 2011,and approved in 2017 with dolutegravir with the drug named Juluca for the treatment of HIV-1 infection in adults.
The synthesis of rilpivirine was shown in Scheme 4,using 2-thioxo-2,3-dihydropyrimidin-4(1H)-one as the starting material.Intermediate 2-(methylthio)pyrimidine-4(1H)-ones 40 was easily prepared byS-alkylation of thiouracil 39 with methyl iodide in the presence of sodium hydroxide.Intermediate 40 was condensed with 4-cyanoaniline at 180–190 °C for about 8 h under solvent-free conditions to provide 4-(4-oxo-1,4-dihydropyrimidin-2-ylamino)benzonitriles 41,which was chlorinated to 4-(4-chloropyrimidin-2-ylamino)benzonitriles 42viarefluxing with POCl3for 30 min [61].Substitution of intermediate 42 with 4–bromo-2,6-dimethylaniline at 150 °C for 1 h gave the aryl bromide 43.Finally,the Heck reaction between the aryl bromide 43 and acrylonitrile afforded the corresponding rilpivirine 3B [62].
5.Vericiguat (VerquvoTM)
Vericiguat 4,also named BAY 1021189,was developed by Bayer and Merck as a soluble guanylate cyclase (sGC) stimulator for treating the disease of chronic heart failure [63,64].Vericiguat is also an important enzyme in nitric oxide signaling pathway.When nitric oxide binds to guanylate cyclase,vericiguat catalyzes the synthesis of intracellular cyclic guanosine phosphate.Vericiguat is a class II drug of biopharmaceutical classification system with low solubility,low clearance,high permeability,and weak alkalinity [65].Vericiguat contains a fluorinated 1H-pyrazolo[3,4-b]pyridine unit,a fluorinated phenyl ring,a pyrimidine-triamine,and a methyl carbamate moiety (Fig.5).The SAR studies by Bayer showed that the introduction of an additional fluorine atom in vericiguat led to good activity.Good bioactivity was observed with MEC value of 0.3 μmol/L compared with 0.7 μmol/L of the analog 44 with one fluorine substitution (MEC: minimal effective concentration to achieve stimulation of cGMP formation in a recombinant sGCoverexpressing cell line) [66,67].Vericiguat was first approved by the FDA in 2021 with the trade name as Verquvo for use in patients with symptomatic chronic heart failure (HF) to reduce the risk of cardiovascular death and HF hospitalization [63,68].
Bayer developed a synthetic strategy for the preparation of vericiguat 4 with 2,2,3,3-tetrafluoro-1-propanol as the starting material,which was shown in Scheme 5 [66].Initially,2,2,3,3-tetrafluoro-1-propanol 45 was activated by trifluoromethanesulfonic anhydride and followed by the treatment with morpholine at 5 °C to give the fluoroalkylated morpholine 46 in 84% yield.The intermediate 46 was methylated with methyl methanesulfonate at 135 °C to yield the quaternary ammonium intermediate 47,which underwent an elimination reaction in the presence of sodium hydroxide.The salt 48 was converted to unsaturated aldehyde 49viatreatment with morpholine and triethylamine.Acrolein derivative 49 reacted with ethyl 5-amino-1-(2-fluorobenzyl)-1Hpyrazole-3-carboxylate 50 in the presence of MsOH and LiCl in refluxing ethanol to form the ester intermediate 51 in 81% yield.Ester 51 was converted into amide 52viathe reaction with formamide,and dehydrated with POCl3to yield the nitrile 53,which was further converted into amidine 54.The amidine 54 reacted smoothly with (E)-2-(phenyldiazenyl)malononitrile 55 in the presence of trimethylamine to afford the intermediate 56 featuring a pyrimidine ring.Intermediate 56 was subjected to a Pd-catalyzed reduction reaction under a hydrogen atmosphere in DMF,then reacted with methyl chloroformate to give the target vericiguat 4.
6.Asciminib (ScemblixTM)
The BCR-ABL1 oncoprotein is responsible for the progression of chronic myelogenous leukemia (CML).The drug resistance issue is related to the available tyrosine kinase inhibitor therapy targeting ATP-binding site of BCR-ABL1 in patients with Philadelphia chromosome-positive (Ph+) CML.Asciminib 5 was developed by Novartis pharmaceuticals as a potent and selective allosteric ABL1 inhibitor.The US FDA has approved asciminib as an oral drug for the treatment of Philadelphia chromosome-positive chronic myeloid leukemia for patients with CML and Ph+ acute lymphoblastic leukaemia in October 2021 [69–71].This compound may also treat or prevent non-malignant diseases associated with abnormally activated ABL1 kinase enzyme [72–74].
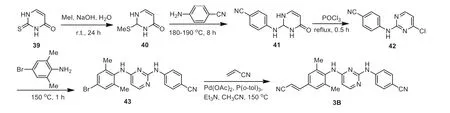
Scheme 4.Synthesis of rilpivirine 3B.
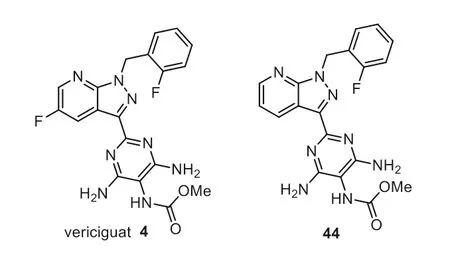
Fig.5.Structure of vericiguat and analog.
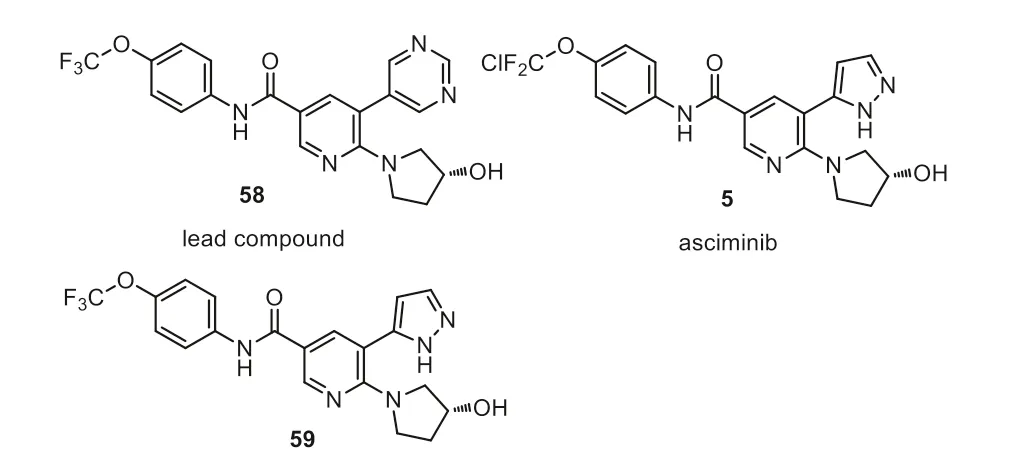
Fig.6.Structure of asciminib and analogs.
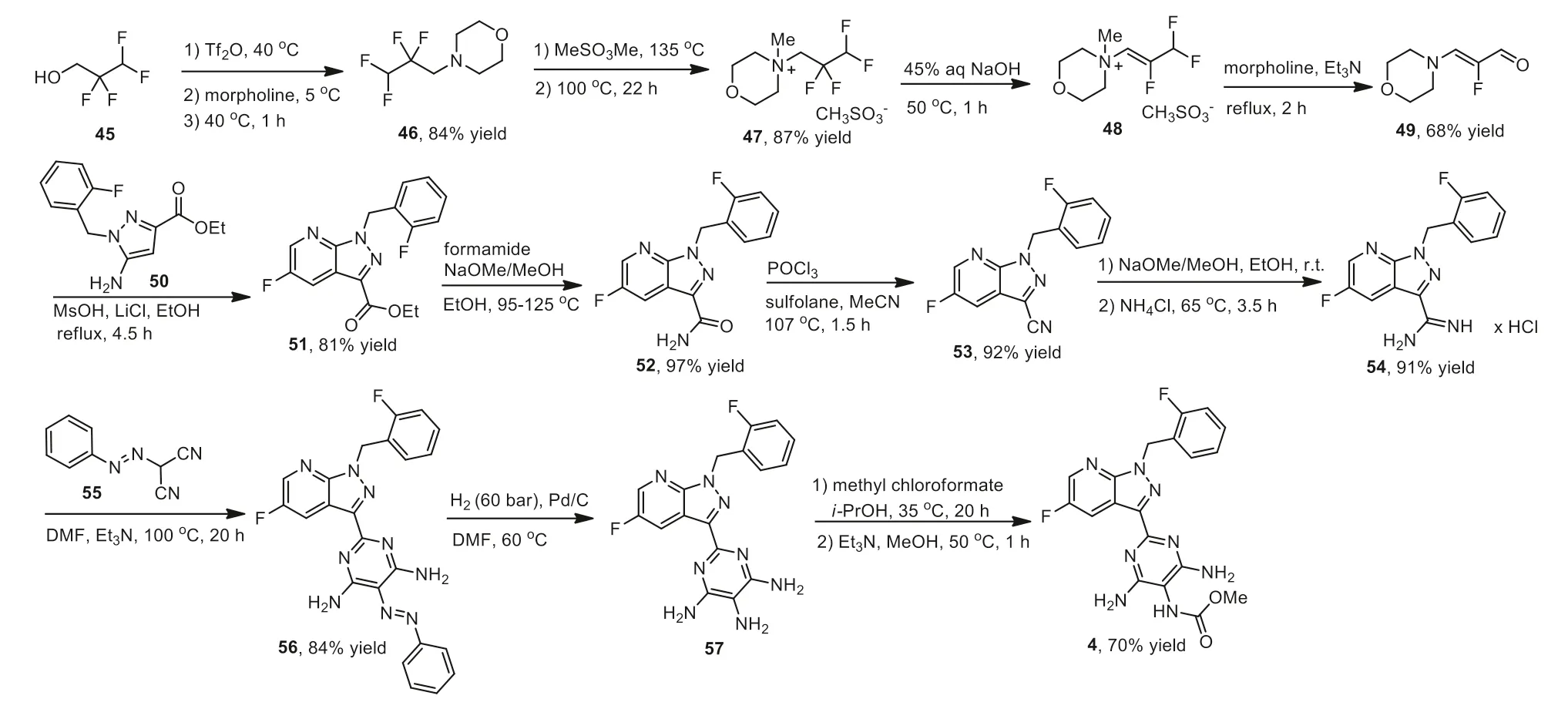
Scheme 5.Synthesis of vericiguat 4.
Asciminib has a unique CF2ClO- group in its structure (Fig.6).The SAR study showed that ap-CF3O- group on the phenyl ring is important for the biological activity (58,ABL1 IC50=2.3 nmol/L)[75].The X-ray co-crystal structure analysis showed that the CF3Omoiety sits in the myristate pocket in an orthogonal position with respect to the plane of the phenyl ring and the fluorine atom interacts with the carbonyl carbon of leucine-359.Those analogs have increased potency in biochemical and cellular assays.Introducing hydroxyl group in the pyrrolidine ring increased the solubility,but decreased the cellular activity.The pyrimidine ring (58,ABL1 IC50=2.3 nmol/L) closed to the backbone carbonyl group of glutamic acid-481 was replaced with 5-membered heterocyclic rings such as pyrazole afforded asciminib 5 showing an IC50value of 0.5 nmol/L.Molecular modeling studies showed that the bulkier group can easily be accommodated by either replacing the oxygen atom with a sulfur atom or replacing one of the fluorine atoms with a chlorine (59,ABL1 IC50=1.1 nmol/L) [76].
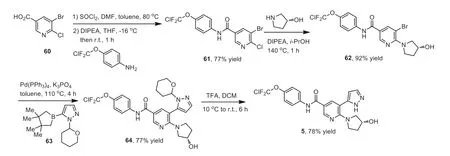
Scheme 6.Synthesis of asciminib 5.
The synthetic route for asciminib 5 was shown in Scheme 6[76].5-Bromo-6-chloronicotinic acid (60) was converted into the acid chloride by the reaction with SOCl2in DMF at 80 °C for 1 h.Then,the concentrated reaction mixture was treated with 4-(chlorodifluoromethoxy)aniline in tetrahydrofuran (THF) in the presence ofN,N-diisopropylethylamine (DIPEA) at room temperature for 1 h to afford compound 61 in 77% yield.Compound 61 reacted with the enantiomerically pure suspension of (R)-pyrrolidin-3-ol ini-PrOH in the presence of DIPEA at 140 °C for 1 h to afford bromonicotinamide 62 in 92% yield.The Suzuki-Miyaura coupling of 62 with protected boronic ester 63 in the presence of Pd(PPh3)4and K3PO4at 110°C for 4 h afforded the tetrahydropyranyl-protected pyrazole analog 64,which was then deprotected with TFA at 10–25 °C for 6 h to provide asciminib 5 in 78% yield.It should be mentioned that the starting 4-(chlorodifluoromethoxy)aniline is commercially available.
7.Atogepant (QuliptaTM)
Calcitonin gene-related peptide (CGRP) is a potent naturally occurring 37-amino acid neuromodulatory peptide localized in the central and peripheral nervous system,which is responsible for several biological actions,including vasodilation.Studies have shown that CGRP receptors are expressed in regions in the brain associated with migraine pathophysiology and CGRP levels are elevated during migraine attacks.It exerts its biological response by binding to specific cell surface receptors coupled to the activation of the adenylyl cyclase enzyme [77–79].In clinical trials,the CGRP antagonist is effective in treating acute attacks of migraine and other neurogenic inflammation and inflammatory pain [80,81].Several labs have reported that the vascular effects of CGRP can be reduced or reversed by a CGRP antagonist [82–87].Merck conducted a SAR study leading to the identification of atogepant 6,as a potent and selective CGRP antagonist.It is the first orally administered calcitonin gene-related peptide (CGRP) antagonist,approved by the US FDA in September 2021 for the prevention of episodic migraine and cluster headache.As shown in Fig.1,two fluorinated groups,trifluorophenyl and trifluoromethyl,are presented in atogepant 6.
Shown in Scheme 7 was the synthesis of atogepant 6 using fluorinated 2-phenylacetic acid 65 as a starting material[88,89].Acid 65 in DMF and isopropyl acetate (iPAc) was converted to acid chloride by reacting with POCl3at 0 °C for 30 min.The resulting mixture was added into a solution of K2CO3and NHMe(OMe)·HCl in water below 8°C to afford 66 in 99%yield.The CeCl3promoted reaction between intermediate 66 and MeMgCl in THF,followed by treatment with hydrochloric acid (2 mol/L) and methyltert–butyl ether (MTBE) at 5–10°C to give L-(2,3,6-trifluorophenyl)propan-2-one (67) in 95% yield,which was subjected to a substitution reaction with isopropylN-(tert–butoxycarbonyl)-O-(methylsulfonyl)serinate (68) in the presence of ZnBr2andtBuOLi affording the intermediate 69 in 67% yield after 24 h.Asymmetric cyclization reaction of intermediate 69 in the presence of sodium tetraborate decahydrate,iPrNH2,pyridoxal-5-phosphate (PLP) and SEQ ID No.1 in borate buffer at 55°C for 24 h afforded the carbamate intermediate 70 as a mixture ofcisandtrans-isomers (71% yield).The crude mixture of 70 in Me-THF was treated withtBuOK at room temperature for 2 h and crystalized iniPAc/heptane at 60°C to afford crystalizedcis-70 in 85% yield.The introduction of a trifluoroethyl group with the use of trifluoroethyl trifluoromethanesulfonate and followed by removal of Boc group generated the free amine 71 in 92% yield.Compound 71 coupled with carboxylic acid 72 in the presence of HOBt monohydrate and EDC hydrochloride at room temperature for 4 h giving compound 6 in 95% yield as monohydrate.
8.Avacopan (TavneosTM)
The complement system plays a crucial role in the clearance of immune complexes and the disproportionate activation of the complement system can be manifested in various disorders leading to severe inflammation and tissue damage [90–93].The anaphylactic complement C5a is the most potent inflammatory mediator acting via its interaction with the membrane-bound C5a receptor (C5aR) [94–99].Several labs have reported non-peptide-based C5a receptor antagonists [100,101].The pathogenesis of ANCAassociated vasculitis is characterized by neutrophil based inflammation of small vessels,often including those in the kidney.It is reported that the activation of the alternative complement pathway plays a role in disease pathogenesis [102–104].The US FDA approved avacopan 7 in October 2021,for the treatment of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis.Avacopan is a small molecule developed by ChemoCentryx,antagonizes complement-dependent inflammation through its binding to the C5aR [105].It has two phenyl rings containing one fluorine and one trifluoromethyl group (Fig.7).The SAR studies disclosed that the fluoro and trifluoromethyl substitutions were very important for the improvement of bioactivity.They found that C5aR IC50values for compounds 73A and 73B were both between 5 and 50 nmol/L,while less than 5 nmol/L was observed for avacopan 7[106].
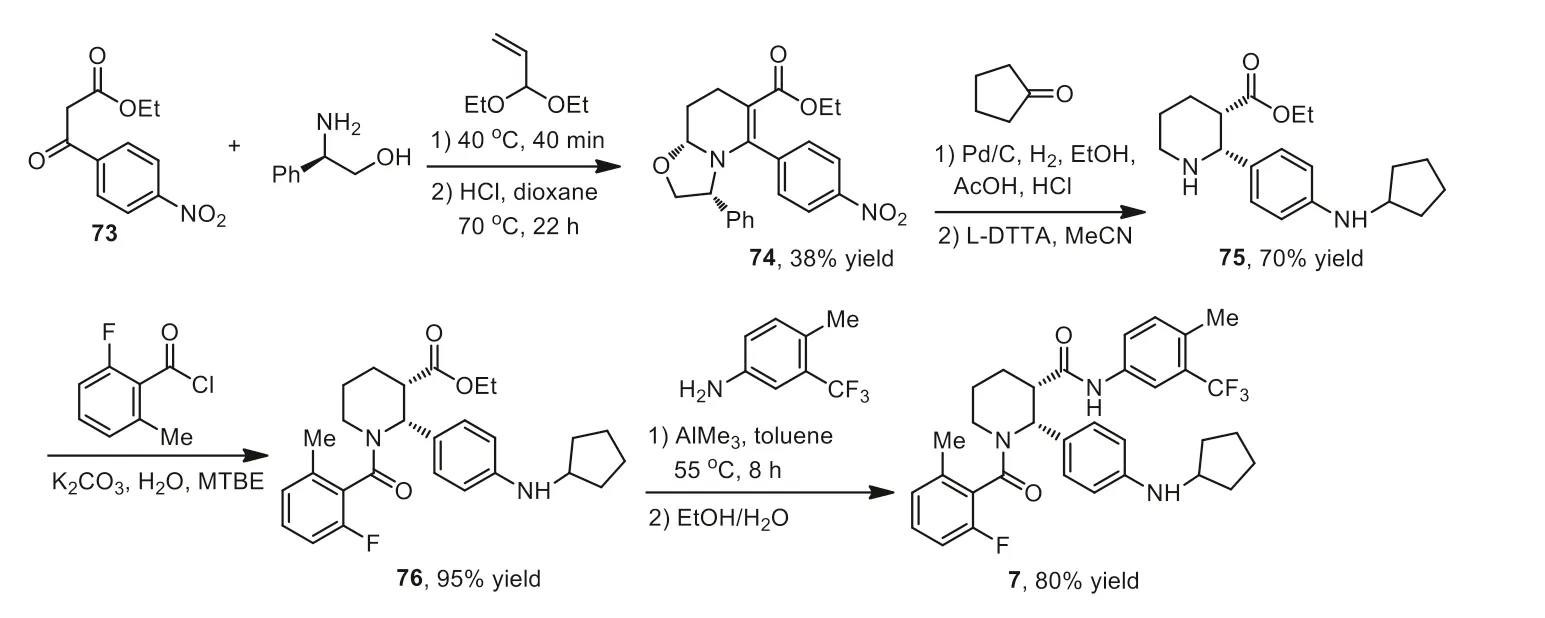
A reported synthetic method for the preparation of avacopan 7 was shown in Scheme 8 [107].Three-component condensation reaction of ethyl 3-(4-nitrophenyl)-3-oxo-propanoate (73),(R)-2-amino-2-phenylethan-1-ol and acrolein diethyl acetal at 40°C,followed by the treatment of 4 mol/L HCl in dioxane at 70 °C for 22 h generated chiral compound 74 in 38% yield.Reductive amination of compound 74 with cyclopentanone,10% Pd/C in ethanol in the presence of glacial acetic acid and 12 mol/L HCl,followed by treatment with di-p-toluoyl-L-tartaric acid (L-DTTA) provided the intermediate 75.Amide coupling of 75 with 2-fluoro-6-methylbenzoyl chloride in the presence of K2CO3in water and MTBE as co-solvent afforded the amide 76 in 95% yield.Finally a solution of 4-methyl-5-trifluoromethylaniline and amide 76 in toluene was treated with AlMe3at 55°C for 8 h to provide avacopan 7,which was recrystallized from ethanol/water to give avacopan 7 as white crystals in 80% yield.
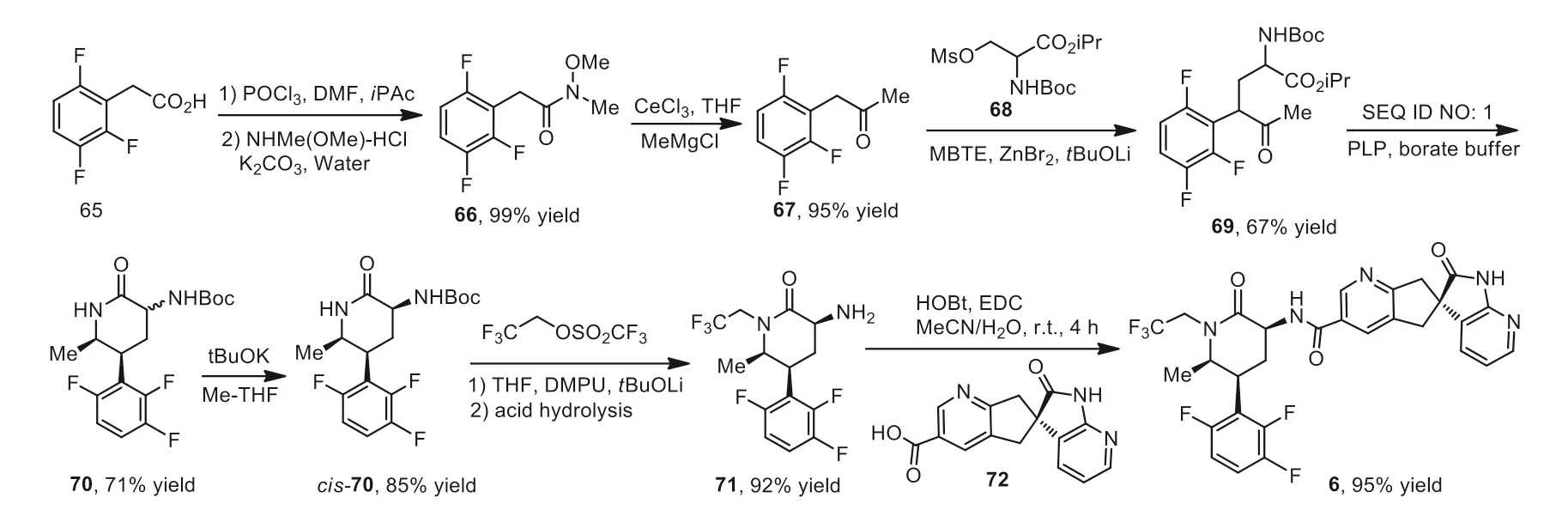
Scheme 7.Synthesis of atogepant 6.
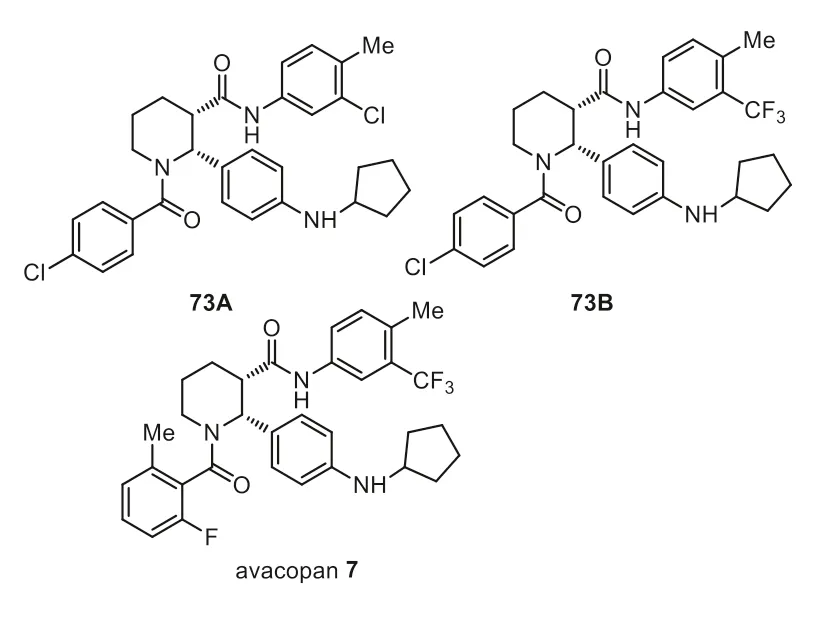
Fig.7.Structures of avacopan and analogs.
9.Belzutifan (WeliregTM)
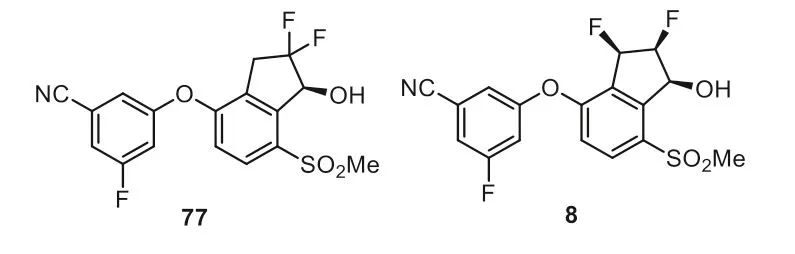
Fig.8.Structure of belzutifan and analog.
The von Hippel-Lindau (VHL) disease is a rare genetic disorder associated with a high risk of multiple organ cancer development[108,109].Renal cell carcinoma (RCC) is characterized by the inactivation of the tumor suppressor VHL protein,a component of an E3 ubiquitin ligase complex responsible for protein degradation.A major role of VHL protein is the regulation of the hypoxiainducible factors (HIF).The hypoxia-inducible factor 2α(HIF-2α)is a critical factor in renal cell carcinoma (RCC).Oxygen-dependent HIF prolyl-hydroxylase enzyme controls the cellular activity of HIF-2α[110,111].In the presence of oxygen,these enzymes hydroxylate specific proline residues to provide a substrate recognition site for VHL protein and target it for rapid proteasomal degradation.The VHL protein is defective or absent in patients with RCC which leads to the accumulation and transcriptional activation of HIF-2α.
A number of research groups have reported that the inner pocket in HIF-2αcould be allosterically inhibited by small molecules and thus the reduced protein-protein interaction between HIF-2αand ARNT leads to the inhibition of transcriptional activity [112–115].The first generation HIF-2αinhibitor 77 developed through structure-guided studies have shown clinical activity in advanced RCC patients [116] but its use was restricted due to its extensive phase 2 metabolismviaglucuronide conjugation [117–119].The structural modification of 77 to belzutifan 8 by altering its geminal difluoro group to a vicinal difluoro group resulted in enhanced potency,decreased lipophilicity,and better ADME profile with reduced formation of the glucuronide conjugate (Fig.8).The US FDA has approved a hypoxia-inducible factor-2α(HIF-2α)inhibitor,belzutifan (Welireg) developed by Merck),for the treatment of cancers associated with VHL disease that does not require immediate surgery.As shown in Fig.8,belzutifan 8 is a chiral compound featuring three continuous chiral carbon centers,which also contains three fluorine substitutions,two C(sp3)-F and one C(sp2)-F.
Scheme 8.Synthesis of avacopan 7.
Scheme 9.Synthesis of belzutifan 8.
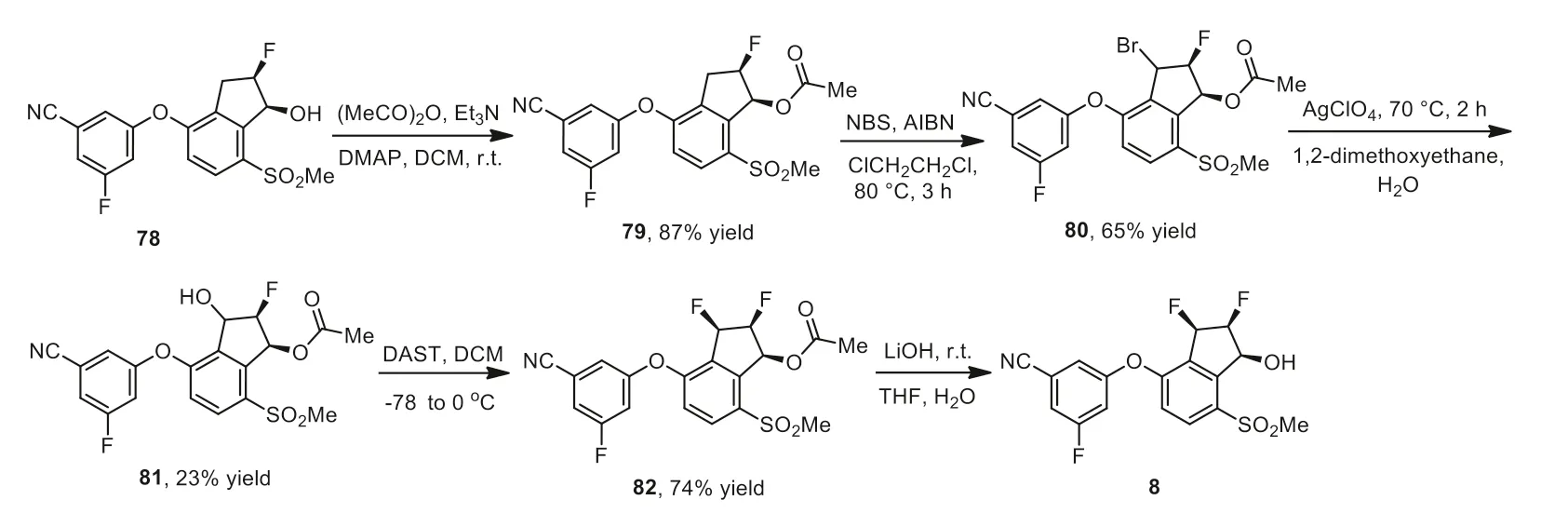
The synthesis of belzutifan 8 was outlined in Scheme 9,which used optically pure fluorinated compound 78 as a starting material [120–122].The alcohol 78 was converted to acetate ester 79 in 87% yield by reaction with acetic anhydride in the presence of 4-(dimethylamino)pyridine (DMAP) and trimethylamine.The ester 79 was then brominated by a radical reaction initiated by azodiisobutyronitrile (AIBN) with the use ofN-bromosuccinimide (NBS)at 80°C for 3 h resulting in the compound 80 in 65% yield.The treatment of 80 with AgClO4at 70°C for 2 h gave the alcohol 81 in 23% yield.Subsequently,stereoselective fluorination reaction of 81 with (diethylamino)sulfur trifluoride (DAST) at -78°C gave the vicinal difluoro compound 82 (74% yield).Finally,the ester group in compound 82 was hydrolyzed by 0.5 mol/L LiOH solution at 0°C to afford belzutifan 8.
10.Sotorasib (LumakrasTM)
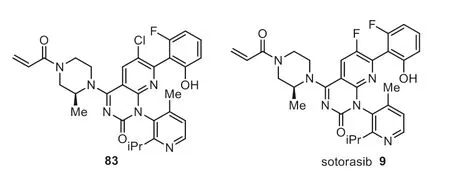
Fig.9.Structure of sotorasib and analog.
RAS is a family of three ubiquitously expressed small GTPases in all animal cell types.Many RAS-driven human tumors are associated with the mutations in the RAS gene family.Of the various isoforms like KRAS,HRAS,and NRAS,the KRAS mutation is the most frequently occurring.The KRAS protein is a signaling molecule capable of downstream regulation of the proliferation of tumors,and the mutations mostly occur in the codon 12 like p.G12D (41%),p.G12V (28%),and p.G12C (14%) [123–126].A series of compounds have been explored as KRAS p.G12C inhibitors interacting with the cryptic pocket of the target protein(H95,Y96,and Q99) [127].Among them,quinazolinone analogs have demonstrated good biochemical and cellular potency.The isopropylphenyl moiety of quinazolinones bonded perfectly with the crucial residues of the cryptic pocket.Molecular docking suggested that the fluorophenol moiety occupying a hydrophobic pocket region could form hydrogen bonding to Arg68 to enhance the potency and MDCK permeability [128–130].Sotorasib 9 was developed by Amgen as a selective and irreversible covalent inhibitor b that interacts with the sulfur atom of the cysteine residue present in the KRas mutant known as KRas G12C [131].Sotorasib 9 contains two C(sp2)-F moieties on different aromatic rings (Fig.9).SAR studies by Amgen showed that replacing the C6 chloro substituent of compound 83 with a fluoro substituent overcame the consistently low bioavailabilities although a modest loss of activity in cellular assays was observed (p-ERK IC50=68 nmol/L and 11 nmol/L for sotorasib 9 and 83 respectively) [130].The US FDA grants accelerated the approval of sotorasib in May 2021 for KRAS G12C mutated NSCLC with the trade name LumakrasTM[131].
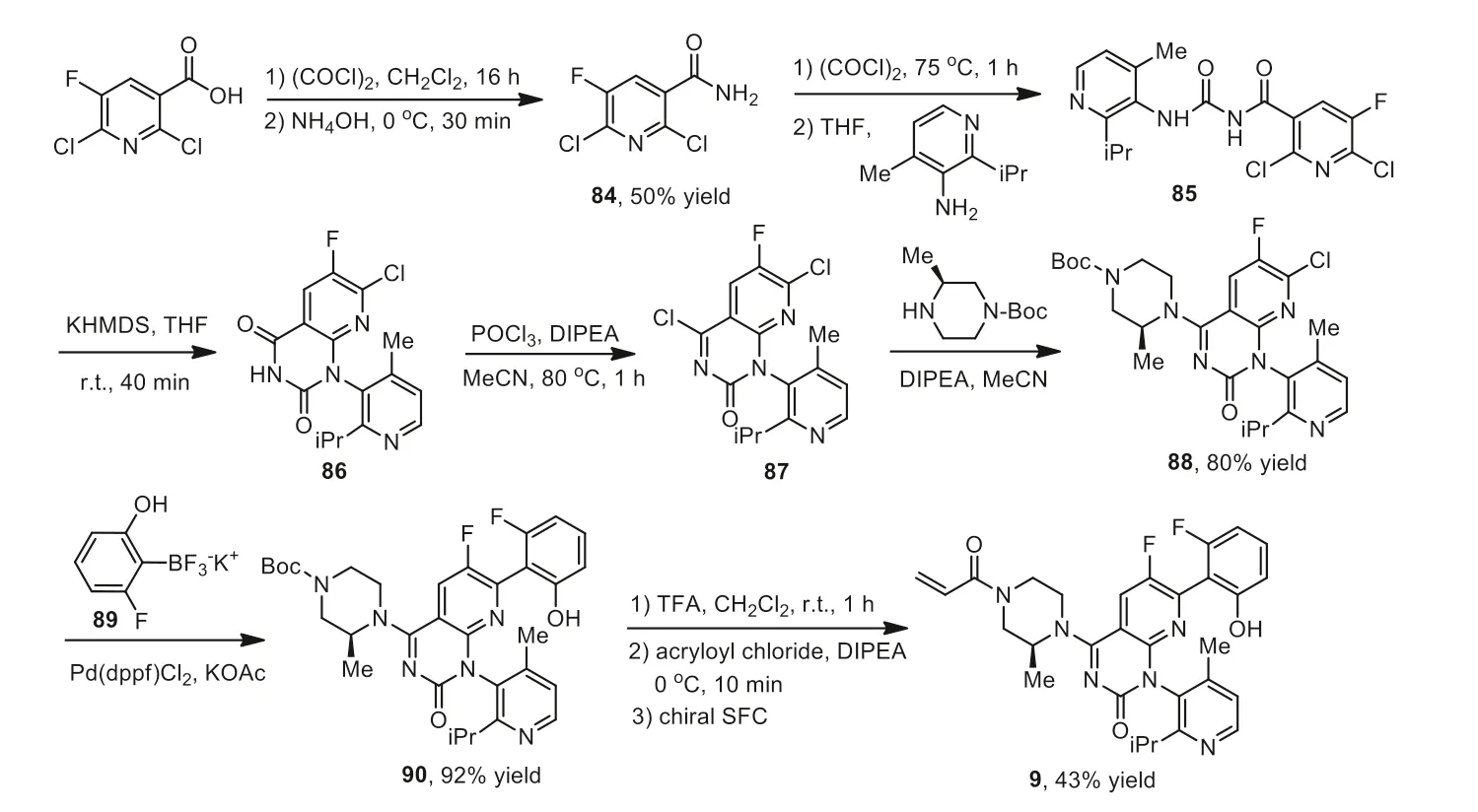
Scheme 10.Synthesis of optically pure sotorasib 9.
The synthesis of sotorasib 9 using 2,6-dichloro-5-fluoronicotinic acid,as a starting material was shown in Scheme 10 [130,132].2,6-Dichloro-5-fluoronicotinic acid was converted to acid chloride by reacting with oxalyl chloride in DCM for 16 h,which then reacted with NH4OH in dioxane at 0°C for 30 min to give 50% yield of nicotinamide 84.Treatment of 84 with oxalyl chloride 75°C for 1 h and followed by reaction with 2-isopropyl-4-methylpyridin-3-amine at 0°C for 1 h yielded the intermediate 85,which was subjected to an intramolecular cyclization reaction in the presence of 1,1,1,3,3,3-hexamethyldisilazane potassium salt (KHMDS) at room temperature to afford cyclic intermediate 86.Compound 86 was chlorinated with POCl3in the presence of DIPEA at 80°C to the chlorinated intermediate 87 which was used without further purification.Reaction of 87 by (S)-4-Boc-2-methyl piperazine with DIPEA as a base afforded 88 in 80% yield.Pd-catalyzed coupling of 88 with potassium trifluoroborate salt 89 in the presence of KOAc afforded 90 in 92% yield.Compound 90 was deprotected via treatment with TFA,acrylation with acryloyl chloride in the presence of DIPEA,and chiral separation by chiral supercritical fluid chromatography (SFC) purification afforded the targeted 9 in 43% yield.
11.Conclusions and outlook
The fluorine editing of drug-candidate molecules is regarded as one of the most promising strategies in the development of modern pharmaceuticals.Last year the FDA approved fifty new small-molecules drugs among which,nine compounds featured fluorine-containing groups.These include Pepaxto (1),Ukoniq (2),Cabenuva (3),Verquvo (4),Scemblix (5),Qulipta (6),Tavneos (7),Welireg (8) and Lumakras (9),representing such therapeutic areas as multiple myeloma,lymphoma,HIV,chronic heart failure,chronic myeloid leukemia,(ANCA)-associated vasculitis,migraines,von Hippel-Lindau disease,non-small cell lung cancer,respectively.This review has covered the aspect of biological activity and the most significant synthetic approaches for the preparation of these new pharmaceuticals.As compared with previous years [26–28],one can notice a significant increase of compounds with multiple fluorine substitutions,including aliphatic (5,6,8)/aromatic (1,2,4,7,9) as well as different types of aromatic fluorine on the same molecule (6,7).This trend indicates a departure from traditional fluorine editing which usually involved a single fluorination.Indeed,the continuous development of fluorine methodology and more deep understanding of the fluorine effect on bio-properties would probably provide strong support for this trend in the future.
Another striking feature of this new crop of drugs is that virtually all,except for (4),are chiral compounds featuring up to three stereogenic centers.Considering the necessity of application of chiral drugs in enantiomerically pure form,critical improvements in asymmetric synthesis and characterization of chiral fluorine-containing compounds have to be made.Thus,studying the self-disproportionation of enantiomers (SDE) properties of these chiral drugs is an important issue,which is recommended in the fluorine-containing drug manufacture [133–138].
Declaration of competing interest
The authors declare no conflicts.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No.21761132021),the Qing-Lan Project of Jiangsu Province (for Han) and IKERBASQUE,Basque Foundation for Science (for Soloshonok).
 Chinese Chemical Letters2023年1期
Chinese Chemical Letters2023年1期
- Chinese Chemical Letters的其它文章
- Diabetic wound healing activated by supramolecular cascade reaction
- MBenes: Two-dimensional transition-metal borides with ordered metal vacancies
- Wet-adhesive materials of oral and maxillofacial region: From design to application
- Diverse catalytic systems for nitrogen-heterocycle formation from O-acyl ketoximes
- The development and application of dual-comb spectroscopy in analytical chemistry
- Boron: A key functional component for designing high-performance heterogeneous catalysts