
桃果实单果重及可溶性固形物含量的全基因组选择分析
2023-03-14 05:39曹珂陈昌文杨选文别航灵王力荣
中国农业科学 2023年5期
曹珂,陈昌文,杨选文,别航灵,王力荣
桃果实单果重及可溶性固形物含量的全基因组选择分析
曹珂,陈昌文,杨选文,别航灵,王力荣
中国农业科学院郑州果树研究所,郑州 450009
【背景】桃单果重和可溶性固形物含量(SSC)是育种家关注的两个重要的数量性状,受到多个微效基因的控制,难以通过单个标记进行早期筛选。全基因组选择作为一种新颖的数量性状早期预测工具,在果树上已经有了初步应用,但其在桃上的应用效果以及影响预测准确性的因素仍需要深入探讨。【目的】建立桃单果重和SSC的全基因组选择技术,为桃高效分子育种技术体系的建立奠定基础。【方法】以520株训练自然群体为试材,通过重测序筛选出的48 398个SNP进行分型,在11个全基因组预测模型中分别筛选出两个数量性状适宜的模型,进而在56株自然群体和1 145株杂交群体上进行应用。【结果】3类群体的平均测序数据量在1.95—3.52 Gb,测序深度为5.29—10.79×。训练自然群体经与参考基因组比对,共得到5 065 726个SNP,去除缺失率较高(>20%)、最小等位基因频率过低(<0.05)的位点后,随机挑选基因组上48 398个SNP用于训练群体的全基因组选择模型构建。单果重预测精度最高的模型是BayesA,SSC预测精度最高的模型为randomforest。分别利用两个数量性状最适的模型进行预测,发现在自然群体中,单果重的预测精度为0.4767—0.6141,高于SSC的0.3220—0.4329;而在杂交群体中,单果重的预测精度为0.2319—0.4870,同样高于SSC的0.0200—0.2793;该结果也表明利用训练自然群体构建的预测模型在预测自然群体上应用的精度高于杂交群体。进而以单果重为例,发现当育种目标是大果时,全基因组选择仅需保留17.78%的单株,效率明显高于单标记和双标记筛选。同时探讨了群体离散程度、遗传力和群体结构等对预测精度的影响,发现预测精度可能受到上述因子的综合影响。【结论】本研究筛选出桃果实单果重和SSC适宜全基因组选择模型,表明该方法的选择效率明显高于单标记筛选,研究结果为两个数量性状的高效分子辅助育种奠定了理论和技术支撑。
桃;单果重;可溶性固形物含量;全基因组选择;早期预测
0 引言
【研究意义】桃是我国主要果树树种,栽培面积和产量均居世界首位。进入21世纪,我国育种家利用常规育种技术选育出的品种,逐渐取代了20世纪日本、韩国品种在我国的主导地位。然而,多年生果树常规育种费时费力,成为限制桃育种水平进步的障碍。随着分子生物学的发展,分子育种可以有效地提高果树育种效率,成为实现丰产优质、提高贮运性能、增强抗逆性等果树育种目标的重要手段[1]。【前人研究进展】在桃上,已有不少重要农艺性状的优异基因被发掘,且开发出准确的、实用的前景选择分子标记,如与桃果皮毛有/无性状相关的位点为第5染色体上第3个外显子上的一个转座子插入[2],与肉质相关的位点为第4染色体两个串联排列基因和的存在与缺失变异[3],与果形扁/圆共分离的变异为第6染色体上的一个长度为1.67 Mb的染色体倒位[4]。然而,上述性状均为质量性状,而育种家更为关注的性状如单果重、可溶性固形物含量(SSC)和果皮着色等性状多为数量性状,由多个基因控制。如Dirlewanger等[5]利用普通桃种质FerjalouJalousia与油桃Fantasia杂交的F2群体,进行连续两年的农艺性状评价,将单果重定位在第6连锁群。QUILOT等[6]利用山桃种质P1908与黄肉油桃Summergrand杂交的BC2群体,将单果重性状定位在第1、2、4、5和7连锁群。EDUARDO等[7]利用Contender×Ambra群体,在第4和6连锁群获得了年度重复的单果重性状的数量性状位点(QTL)。随后,DA SILVA LINGE等[8]首次采用9 K的SNP芯片,将单果重定位到第1、2、3、5、6和7连锁群。ZEBALLOS等[9]同样利用上述芯片,在连锁群1、4、8上鉴定出了与单果重连锁的QTL。综上,在桃的8个连锁群上,均有单果重QTL定位的报道,根据上述定位结果开发的标记由于遗传背景狭窄、标记的变异解释率较低,因此通用性差,难以在其他群体上进行应用。近年来,全基因组选择作为一种新的目标性状早期预测工具,在果树上有了一定程度的应用,该技术利用覆盖全基因组的高密度分子标记,通过构建预测模型,计算个体的基因组育种值,然后根据育种值的大小进行选择。该方法可以加快育种进程、降低育种成本,对于遗传力较低以及难以测量的复杂性状具有良好的预测效果[10]。该方法在动物[11]遗传育种中发挥了重要作用,在作物[12]和林木[13]上的应用处于初级阶段。在苹果上,KUMAR等[14]利用来自4个母本和2个父本杂交的包含1 120个单株的杂交群体,采用包含8 000个SNP的Infinium SNP芯片进行基因分型,然后采用RR-BLUP和Bayesian LASSO模型进行果实性状的育种值估计。研究发现,两种方法所得到的预测准确性比较相似,均在0.7—0.9。以RR-BLUP方法为例,SSC的预测准确性最高,其次为果实硬度和可滴定酸(TA),最低是食用果肉时的收敛性。研究认为,标记的数量、基因组连锁不平衡水平会影响全基因组选择的准确性。MURANTY等[15]利用包含20个全同胞家系的977个苹果单株为训练群体,以5个全同胞家系的1 390个单株为预测群体,采用BayesCπ模型,分析了全基因组选择对10个果实相关性状的预测效果;结果发现,在这些性状中,平均预测准确度最高的为果皮着色百分比,其次为果皮着色、果实大小、外观鲜艳程度和果皮锈斑等,预测能力较弱的有着色类型、采前落果和裂果率等指标,表型分布的离散程度及遗传力大小是影响全基因组选择预测准确性的重要因素。在桃上,BISCARINI等[16]以来自意大利、法国和西班牙的11个杂交群体的1 147个单株作为训练群体,对单果重、SSC和TA的3—5年的表型数据进行了全基因组选择分析,选择9 K IPSC芯片中的6 076个SNP的分型结果,结合GBLUP模型进行育种值预测。研究发现,在不同群体中,SSC的预测准确性最高,达到0.72,其次是TA的0.65,最低是单果重0.6。研究结果显示,群体数量和表型变异越大,预测的精度越高。在梨上,MINAMIKAWA等[17]选择包含86个品种的自然群体和16个全同胞系的765个单株的杂交群体为材料,完成了18个性状的表型评价,进而利用1 506个SNP的基因型分型结果进行全基因组选择分析,发现当自然群体和杂交群体合并在一起,能够明显提高育种值预测的准确性。在杏上,NSIBI等[18]利用1个包含153个单株的来自杏Goldrich×Moniqui的F1群体进行全基因组选择分析,表型为连续2年的果实相关性状,基因型数据来自简化基因组测序产生61 030个SNP的分型信息。该研究利用6个模型估算10个性状(单果重、乙烯含量、果皮底色、可滴定酸、3种糖组分、2种酸组分以及果形指数)的育种值,发现6个性状中,RR-BLUP模型的预测准确度均高于其他5个模型。研究同时发现,随着训练群体数目从总数的25%增加到75%,预测准确性持续增加;而标记数目从50增加到6 103个(10%的总标记)标记时,预测准确性持续增加;之后,标记数目的增加对预测准确性增幅不大。【本研究切入点】综上,国内外对于果树甚至桃的全基因组选择已有研究,但多以杂交群体为主,仅梨上有利用自然群体的报道。同时,许多研究均发现群体数量和标记数量均会影响全基因组选择的准确性。【拟解决的关键问题】本研究以520个桃自然群体的单株为试材,利用高达50 K的SNP分型结果,以单果重和SSC为例,探讨在桃上进行数量性状全基因组选择的应用效果,并分析影响预测准确性的因素,为桃高效分子育种技术体系的建立奠定基础。
1 材料与方法
1.1 植物材料
1.2 性状评价
参考王力荣[19]的方法,测定3类群体果实成熟期的单果重和SSC,并鉴定了2个质量性状(果形扁/圆和果皮毛有/无);其中,单果重为10个果实的平均值,SSC为3个果实的平均值。自然群体的表型性状分别在2012、2015和2016年进行采集,杂交群体的表型性状则分别在2019、2020和2021年进行采集。利用训练群体进行模型构建时,所用的表型为2012、2015和2016年的平均值。
1.3 基因型评价
3个群体的基因型评价采用相同方法,即取幼嫩叶片,采用CTAB法[20]提取总的基因组DNA,经过琼脂糖电泳质检合格后,参考GenoBaits DNA-seq Library Prep试剂盒的使用说明构建重测序文库,文库质量检测合格后,利用华大MGI-2000/MGI-T7测序平台进行测序,测序模式为PE150模式。测序所得的raw reads,经测序数据质控即使用fastp软件[21]去除接头和低质量的reads后获得clean reads,使用BWA软件[22]将clean reads与参考基因组进行比对,继而使用GATK软件[23]的UnifiedGenotyper模块获得每个个体的SNP信息。
1.4 遗传力和群体结构分析
利用表型数据和筛选后的基因型数据,首先使用R软件包(rrBLUP)的kin.blup函数估算遗传方差(σG2,genetic variance)和误差方差(σE2,error variance),然后估计各性状的遗传力。其公式为:h2=σG2/(σG2+σE2)。
使用GCTA软件[24]基于过滤后的SNP信息,进行主成分分析(PCA),获得各个PC的方差解释率及样本在各个PC中的得分矩阵,从SNP信息中提取的关键信息按照效应从大到小分为PC1、PC2、PC3...,以表征群体结构信息。
1.5 基因组预测模型
本研究采用11种模型,包括rrblup、svmrbf、svmpoly、randomforest、pls、gblupD、gblupA、BayesA、BayesB、BayesC、BayesLasso,对训练群体的基因型数据和表型数据进行训练,并通过5-fold交叉验证计算预测精度,然后对预测群体进行表型值预测。所有模型均使用R语言包实现,其中rrblup使用mix.solve,svmrbf和svmpoly使用kemlab,randomforest使用randomForest,pls使用plf,gblupD和gblupA使用kin.blup函数,BayesA、BayesB、BayesC和BayesLasso均使用BGLR。
另外,5-fold交叉验证具体方法为:将训练群体随机分成5份,其中4份作为训练集,剩余1份作为测试集,每一份抽样均有机会作为测试集,并且计算测试集的真实值与预测值间的皮尔森相关系数。重复交叉验证4次,最终的相关系数均值作为预测精度()。
1.6 预测值和真实值之间的符合度
在训练自然群体中,筛选单果重和SSC性状预测精度最高的模型,然后用该模型估计预测自然群体和杂交群体的基因组育种值。最后,计算预测群体的估计值和真实值得到皮尔森相关系数,用来评价全基因选择模型在桃两个数量性状上的应用效果。
2 结果
2.1 训练和预测群体的重测序
本研究所用的群体共3类。如表1,训练自然群体为520个单株,而预测自然群体数目为56株、预测杂交群体1 145株。单个样本的平均测序数据量在1.95—3.52 Gb,测序深度为5.29—10.79×,与参考基因组比对后,覆盖度为78.22%—89.58%。
为促进幼果快速膨大、减少畸形果与落果、提早上市,可以喷施赤霉酸5~6 mg/L+芸苔素0.02~0.03 mg/L。
2.2 训练群体的基因型鉴定
所有样本共检测出5 065 726个SNP,去除缺失率在20%以上的位点后剩余4 254 657个SNP,继续去除最小等位基因频率小于0.05的位点后剩余3 231 646个SNP,然后采用随机挑选的方法,选择48 398个SNP用于后续训练群体的全基因组选择模型构建。这些SNP在桃不同染色体的分布如表2所示,SNP密度平均每Mb达到214.44个,分布密度最高的为第4染色体,密度最低的为第5染色体。

表1 本研究中所用材料的重测序结果

表2 用于全基因组选择分析的SNP在染色体上的分布
观察SNP在不同染色体上的分布(图1),发现在第2染色体的3—5 Mb、第4染色体的0—1 Mb和23—25 Mb存在密度较高的热点区域,这些密度较高的区域与前人报道的SNP在基因组上的分布[25]一致。
2.3 训练和测试群体中两个数量性状的表型分布
如图2所示,训练群体的两个表型性状为正态分布,呈现数量性状的典型特征。单果重的变异系数为31.43%,高于可溶性固形物的14.20%。
两类预测群体的表型分布如图3所示。在自然群体中,2012与2015及与2016年的重复单株较少,因此没有计算其重复性。2015与2016年间的单果重相关性为0.73,达到显著水平;而SSC的相关性仅为0.16,且不显著。在杂交群体中,2019和2021年的单果重相关性最高,达到0.81(<0.01);2020与2021年相关性也较高,2019与2020年相关性不显著,表明环境对2019年的单果重性状影响较大;而SSC则以2019与2020以及与2021年间显著相关,暗示2021年的SSC性状受环境影响较为明显。
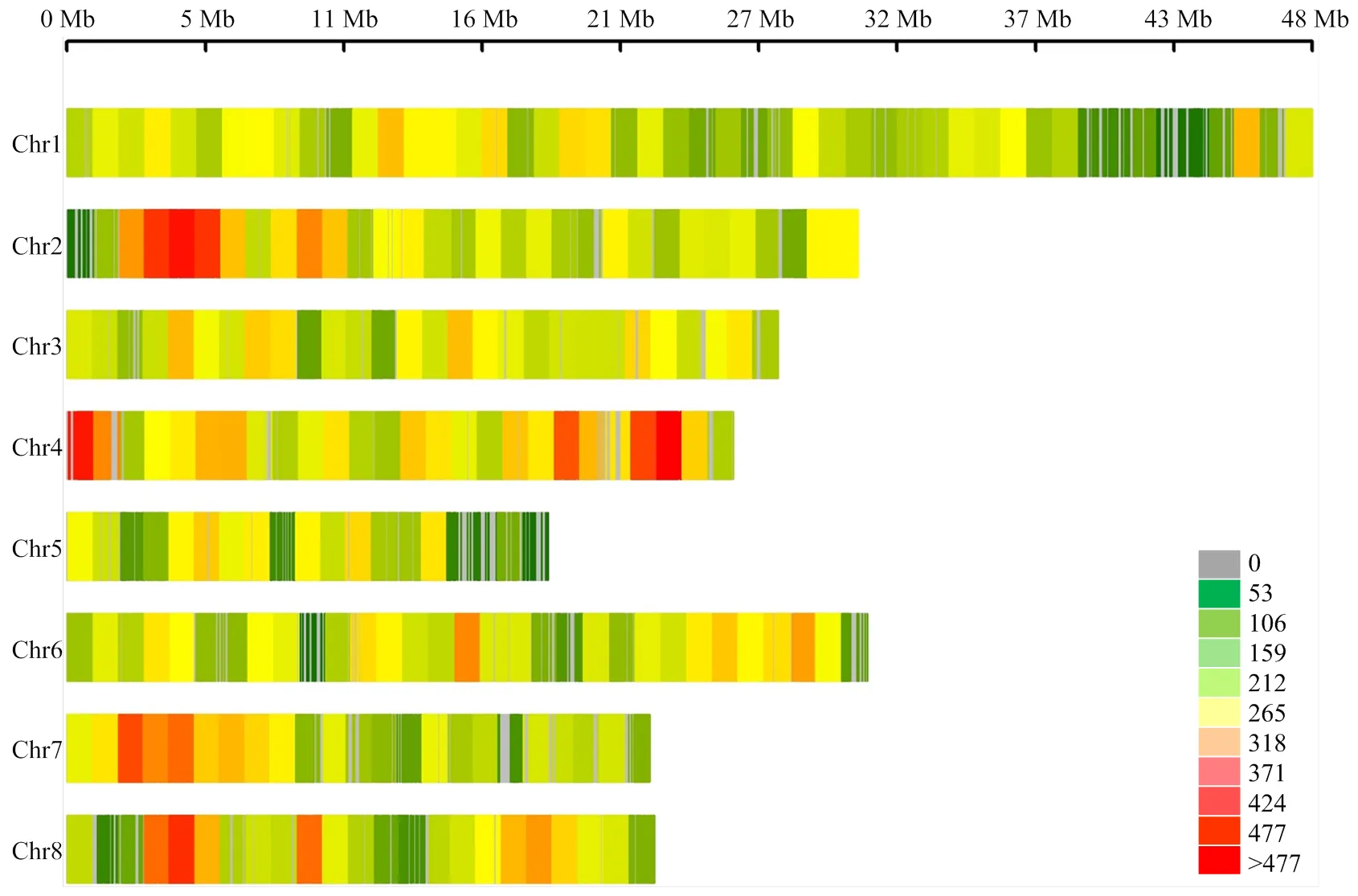
图1 筛选后的SNPs在桃基因组上的分布

图2 训练群体两个数量性状的表型分布

表格中显示为3年性状的相关性The values in the table indicate the correlation between phenotypes evaluated in different years
2.4 基于训练群体的不同预测模型的精度
利用前期鉴定出的48 398个SNP,基于训练群体进行5-fold交叉验证,计算每个模型的预测精度。
如图4所示,单果重性状采用不同模型的预测精度均高于SSC,在11个模型中,单果重预测精度最高的模型是BayesA,精度为0.6017;最低为svmrbf,精度仅为0.4576。SSC预测精度最高的模型为randomforest,精度达到0.4306;最低为svmpoly,精度仅为0.2607。
2.5 用最适宜的模型进行预测群体的育种值估计
分别用最适宜的模型进行两类预测群体的育种值估计,并比较其与真实观测值的相关性。其中,自然群体(图5)3年的单果重表型与预测值的相关性为0.4767—0.6141,高于SSC的0.3220—0.4329,其中单果重以2012年预测最准确,SSC则以2015年的准确度最高。
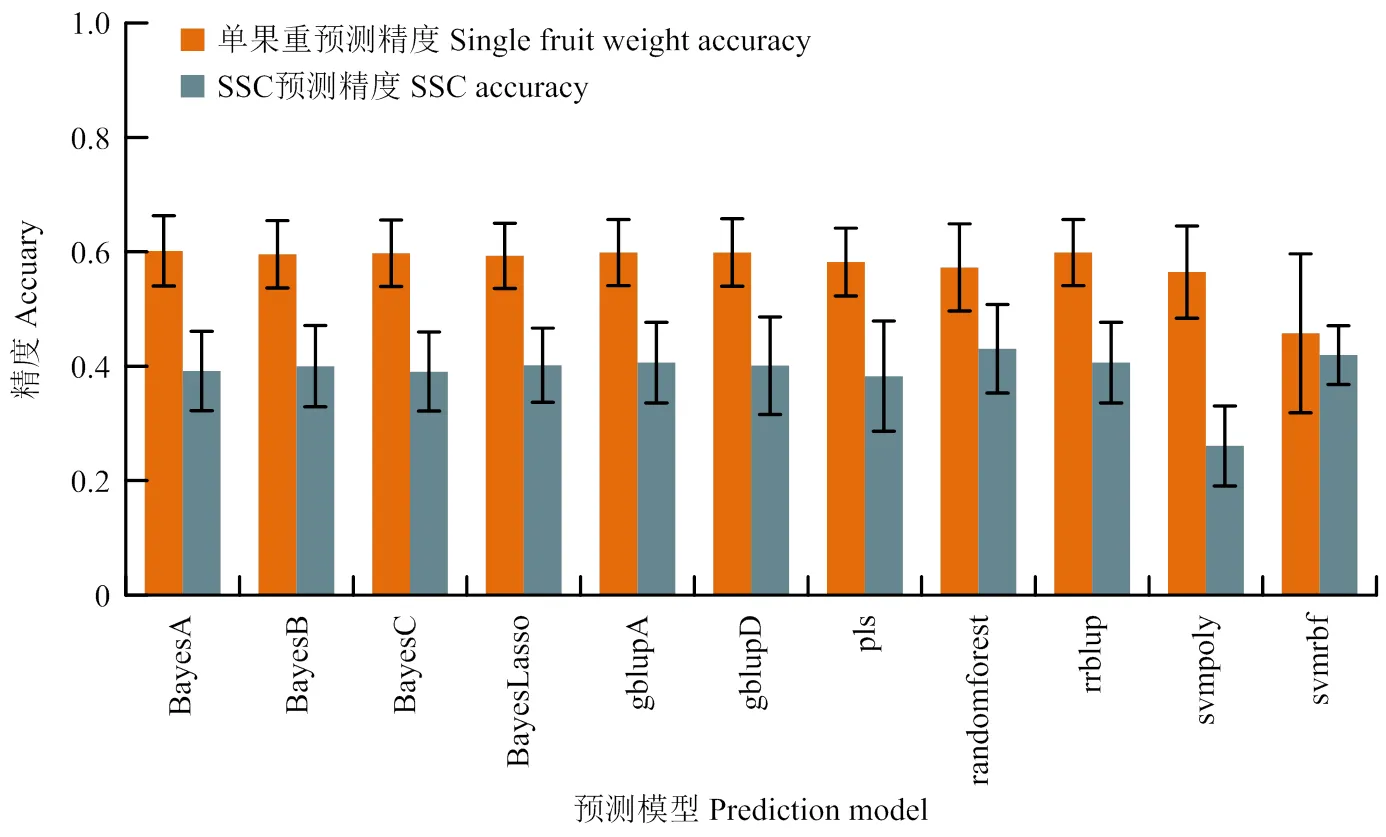
图4 两个数量性状采用不同预测模型的预测精度
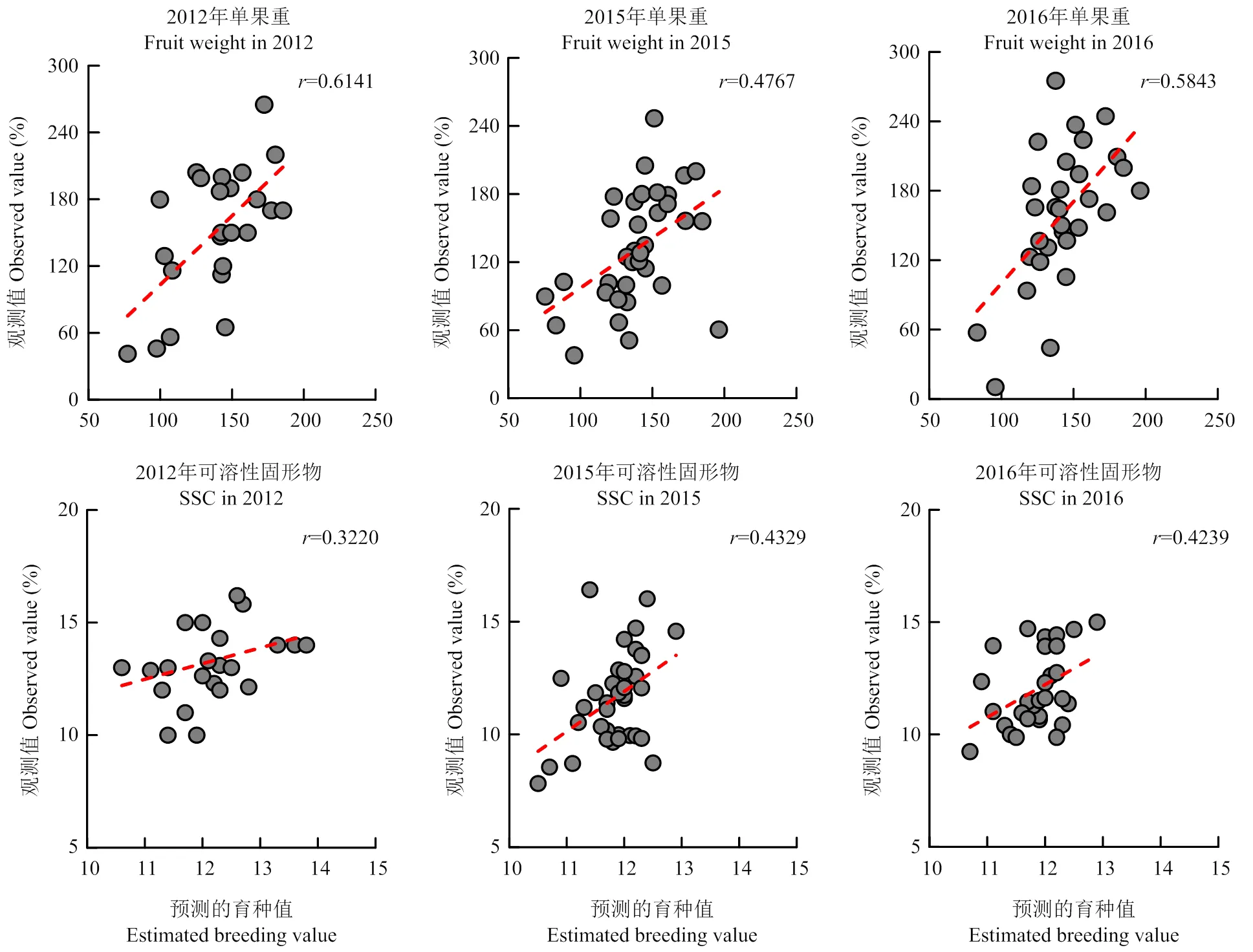
图5 预测自然群体两个数量性状的预测值与观测值的相关性分析
对于杂交群体(图6),研究发现无论是单果重还是SSC,其准确度较自然群体均有所下降,尤其是SSC,不同年份的预测精度低至0.0200—0.2793。
2.6 单果重性状的单标记筛选与全基因组选择的效率比较
由于2019年度的单果重性状进行全基因组选择的精度较高,因此,以该年度数据为例,比较其与单个标记进行筛选的效率。如图7所示,在单标记方法中,本研究使用的是笔者团队前期鉴定出的不同年份关联程度最高的两个标记Chr6: 2 281 398 bp和Chr6: 3 296 344 bp[26],从结果可以看出,Chr6: 2 281 398 bp的CC基因型表型与CG基因型没有差异,而CG基因型与GG基因型的表型差异显著性值为2.5E-07。Chr6: 3 296 344 bp的AA基因型与AG基因型的表型同样无显著差异,而AG基因型与GG基因型的表型差异显著性值达到2.4E-16,即区分效率高于Chr6: 2 281 398 bp。
与单标记难以区分优势等位基因型的杂合与纯合类型不同(如Chr6: 2 281 398 bp的CG和CC,以及Chr6: 3 296 344 bp的AG和AA),根据全基因组选择的预测值可将杂交群体分为3个类群,且类群间的差异均达到显著水平(<0.05)。
进一步将上述两个单果重的分子标记组成9种单倍型,结果发现,7种单倍型(Chr6: 2 281 398 bp和Chr6: 3 296 344 bp分别组成的CG:AG、CC:AA、GG:AG、GG:AA、CG:AA、CC:AG和CC:GG)的单果重较大,但类型间并没有明显差异;单倍型CG:GG的单果重表现为中等,而单倍型GG:GG的单果重最低(图8)。
2.7 性状遗传力对群体预测精度的影响
前人研究认为,性状的遗传力是影响全基因组选择精度的重要因素[27-30],因此,本研究评价了单果重和SSC的遗传力,并以两个质量性状(果形扁/平和果皮毛有/无)为对照,随机选择gblupA模型进行育种值的估计。结果显示,遗传力最高的为果形(0.8185),其次为单果重(0.7021)和果皮毛(0.6866),最低为SSC(0.2815),而预测精度从高到低依次为果皮毛(0.8293)、果形(0.7300)、单果重(0.5986)和SSC(0.4064)。即在两个数量性状间,遗传力较高的性状其表型预测精度较高;但当与质量性状比较时,尽管单果重的遗传力高于果皮毛,但其预测精度却低于后者;同时,在两个质量性状之间,遗传力与全基因组选择的预测精度同样不一致。

图6 预测杂交群体两个数量性状的预测值与观测值的相关性分析

图7 单果重单标记筛选与全基因组选择的效率比较
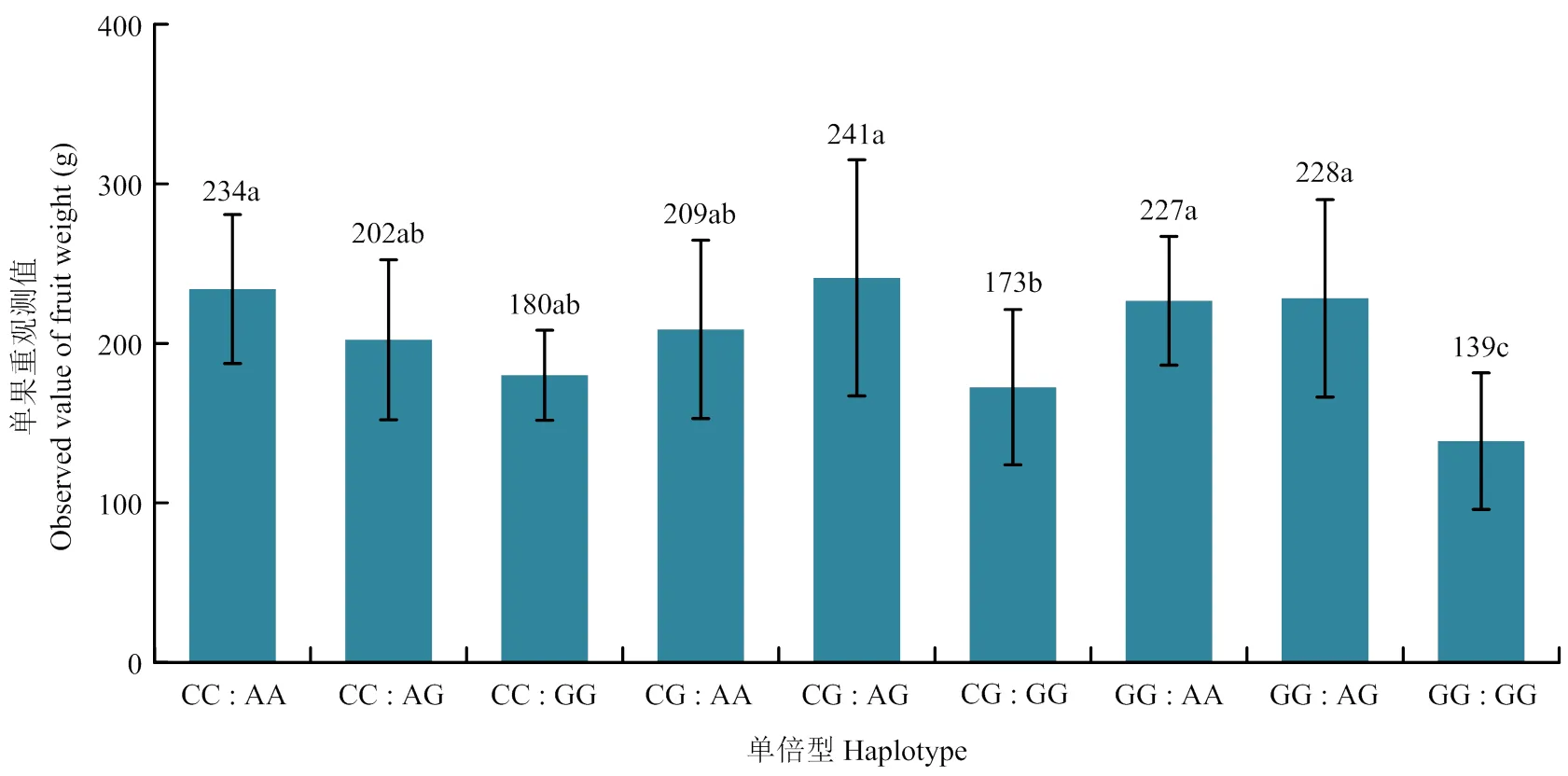
不同小写字母表示处理间差异显著(P<0.05) Different lowercase letters indicate significant differences between treatments (P<0.05)
2.8 群体结构对杂交群体预测精度的影响
群体结构也会影响预测精度[31],在本研究中,以杂交群体表型评价最完善的2020年单果重数据为例,首先进行单个杂交群体的单果重预测育种值与观测值的相关性分析(表3),发现不同群体的相关性差异较大,分布在-0.15—0.26。在相关性为负值的群体中,既有群体大小为27的小群体,也有单株数目达到97的中等群体。
由于本研究是根据自然群体的预测育种值与表型进行比较而筛选精度较高的全基因组选择模型,并进行随后的应用验证。因此,以自然群体为背景,与上述20个杂交群体一起进行PCA分析。如图9所示,相对于其他群体,群体2、4、5和13与背景群体的遗传距离较远,其预测育种值与观测值的相关性较低,分别为0.03、-0.03、-0.15和-0.04;但群体7、11、14、15和19与自然群体的亲缘关系并不远,其预测育种值与观测值的相关性却仍为负值。
3 讨论
3.1 全基因组选择技术在桃上的应用效果
单果重和SSC是桃重要的育种目标性状,研究其早期选择技术体系对其分子育种具有重要的实践意义。理论上,全基因组选择技术将有效地提高数量性状的选择效率。例如本研究比较了单标记和全基因组选择对单果重早期选择的影响,发现当采用单标记时,如以大果为育种目标,则需要保留纯合或杂合的优势等位基因,以应用Chr6: 3 296 344 bp为例,需要保留72株,占整个群体的40.68%;当采用双标记时,需要保留7种单倍型,共71株,占整个群体的42.77%,与采用单标记的应用效率相比没有明显提升。而采用全基因组选择,仅需保留预测育种值最高的一类,约32株,占整个群体的17.78%,筛选效率显著提高。前人研究同样表明全基因组选择的效率高于分子标记辅助选择[32-33]。
3.2 影响全基因组选择效率的因素分析
本研究以单果重和SSC为例,发现单果重预测精度高于SSC,同时利用自然群体构建的模型更适宜预测自然群体。对于杂交群体预测准确率稍低的原因,推测可能是其遗传背景与训练自然群体较远;然而,当比较不同的杂交群体时,却发现预测精度与该单个群体和训练自然群体间的亲缘关系远近无关(图9);对于训练群体与预测群体的遗传相关性对预测精度的影响,需要后续进行更加深入的研究。
对于影响全基因组选择效率的其他因素,前人有不少研究。如MURANTY等[15]在苹果上的研究表明,遗传力大小是影响全基因组选择预测准确性的重要因素。而在桃上,BISCARINI等[16]发现在单果重、SSC 和可滴定酸(TA)共3个性状中,虽然TA的遗传力最高(平均0.53),但预测精度最高的却为SSC(平均0.72)。本研究同样发现,无论是将数量性状与质量性状进行比较或者是比较两个质量性状,全基因组选择的预测精度与遗传力均不呈正相关。同时,MURANTY等[15]和BISCARINI等[16]分别在苹果和桃上均认为表型的离散程度越大,预测的精度越高。本研究中,单果重的变异系数高于SSC,但其是否为单果重性状预测精度高于SSC的主要原因仍有待探讨,笔者更倾向于认为单果重有主效基因而SSC不明显[26],这可能是单果重性状预测精度高的主要原因。此外,在本研究中,由于全基因组选择的预测模型构建基于自然群体3年的平均值,在进行应用时,如果实测的表型值易受环境影响,在理论上会与预测的育种值差异较大。因此,本研究试图探讨全基因组预测精度与表型值在年度间相关性的关系,在预测杂交群体中,尽管2019年的单果重相对其他年份受到环境影响更加明显,然而该年度的表型预测精度却最高,达到0.4870。最后,本研究也探讨了群体结构对预测准确性的影响,同样表明影响全基因组选择精度的因素复杂多样。此外,在前人研究[12]中提到的分子标记密度以及训练群体样本大小等影响全基因组选择准确率的因素,本研究并没有关注,有待开展更广泛深入的研究以归纳总结。

表3 不同杂交群体单果重的预测育种值与2020年观测值的相关性分析
3.3 应用全基因组选择技术对性状的要求
对于全基因组选择的适用范围,本研究在分析遗传力对预测准确性的影响时,加入了两个质量性状,即果皮毛有/无和果形扁/圆,发现这两个性状的预测精度分别为0.8293和0.7300,低于采用单标记进行筛选的准确率(分别为92.86%和100%)[34]。该结果再次表明,全基因组选择对难以测量的复杂性状具有良好的预测效果,对于主效基因和关键变异明确的质量性状,反而过度考虑了其他无效变异的效应,降低了育种值估计的准确性。

图9 共20个杂交群体的PCA分析
4 结论
本研究通过对训练自然群体进行基因分型,筛选了桃果实单果重和可溶性固形物含量(SSC)的适宜全基因组选择模型,并在预测自然群体和杂交群体上进行应用。研究发现单果重预测精度最高的模型是BayesA,SSC预测精度最高的模型为randomforest。在不同模型下,单果重的育种值估计精度均高于SSC。以单果重为例,对数量性状进行全基因组选择的筛选效率明显高于单标记。
[1] 苑兆和, 陈立德, 张心慧, 赵玉洁. 果树分子育种研究进展. 南京林业大学学报(自然科学版), 2021, 45(4): 1-12.
YUAN Z H, CHEN L D, ZHANG X H, ZHAO Y J. Advances in molecular breeding of fruit trees. Journal of Nanjing Forestry University (Natural Science Edition), 2021, 45(4): 1-12. (in Chinese)
[2] VENDRAMIN E, PEA G, DONDINI L, PACHECO I, TERESA DETTORI M, GAZZA L, SCALABRIN S, STROZZI F, TARTARINI S, BASSI D, VERDE I, ROSSINI L. A unique mutation in agene cosegregates with the nectarine phenotype in peach. PLoS ONE, 2014, 9(3): e90574.
[3] GU C, WANG L, WANG W, ZHOU H, MA B Q, ZHENG H Y, FANG T, OGUTU C, VIMOLMANGKANG S, HAN Y P. Copy number variation of a gene cluster encoding endopolygalacturonase mediates flesh texture and stone adhesion in peach. Journal of Experimental Botany, 2016, 67(6): 1993-2005.
[4] GUO J, CAO K, DENG C, LI Y, ZHU G R, FANG W C, CHEN C W, WANG X W, WU J L, GUAN L P, WU S, GUO W W, YAO J L, FEI Z J, WANG L R. An integrated peach genome structural variation map uncovers genes associated with fruit traits. Genome Biology, 2020, 21(1): 258.
[5] DIRLEWANGER E, PRONIER V, PARVERY C, ROTHAN C, GUYE A, MONET R. Genetic linkage map of peach [(L.) Batsch] using morphological and molecular markers. Theoretical and Applied Genetics, 1998, 97(5/6): 888-895.
[6] QUILOT B, WU B H, KERVELLA J, GÉNARD M, FOULONGNE M, MOREAU K. QTL analysis of quality traits in an advanced backcross betweencultivars and the wild relative species. TAG Theoretical and Applied Genetics Theoretische Und Angewandte Genetik, 2004, 109(4): 884-897.
[7] EDUARDO I, PACHECO I, CHIETERA G, BASSI D, POZZI C,
VECCHIETTI A, ROSSINI L. QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect. Tree Genetics and Genomes, 2011, 7(2): 323-335.
[8] DA SILVA LINGE C, BASSI D, BIANCO L, PACHECO I, PIRONA R, ROSSINI L. Genetic dissection of fruit weight and size in an F2peach [(L.) Batsch] progeny. Molecular Breeding, 2015, 35: 71.
[9] ZEBALLOS J L, ABIDI W, GIMÉNEZ R, MONFORTE A J, MORENO M A, GOGORCENA Y. Mapping QTLs associated with fruit quality traits in peach [(L.) Batsch] using SNP maps. Tree Genetics & Genomes, 2016, 12: 37.
[10] 倪海枝, 王引, 颜帮国, 陈方永. 果树基因组辅助育种技术研究现状与展望. 分子植物育种, 2021, https://kns.cnki.net/kcms/detail/46. 1068.S.20210416.1640.009.html.
NI H Z, WANG Y, YAN B G, CHEN F Y. Research status and prospects of genomics-assisted breeding technology in fruit trees. Molecular Plant Breeding, 2021, https://kns.cnki.net/kcms/detail/46. 1068.S.20210416.1640.009.html. (in Chinese)
[11] 张顺进, 寇浩玮, 丁晓婷, 刘贤, 蔡雯雯, 张子敬, 施巧婷, 茹宝瑞, 雷初朝, 黄永震. 全基因组选择技术在反刍动物遗传育种中的研究进展及其应用. 农业生物技术学报, 2021, 29(3): 571-578.
ZHANG S J, KOU H W, DING X T, LIU X, CAI W W, ZHANG Z J, SHI Q T, RU B R, LEI C Z, HUANG Y Z. The research progress and application of genomic-wide selection in ruminant genetics and breeding. Journal of Agricultural Biotechnology, 2021, 29(3): 571-578. (in Chinese)
[12] 刘海岚, 夏超, 兰海. 全基因组选择技术在作物育种中的研究进展. 华北农学报, 2022, 37(增刊): 51-58.
LIU H L, XIA C, LAN H. The research progress of genomic selection in breeding of crops. Acta Agricultrae Boreali-Sinica, 2022, 37(Suppl): 51-58. (in Chinese)
[13] 张苗苗, 王军辉, 卢楠, 麻文俊, 王楠, 吴夏明. 林木全基因组选择研究现状和应用. 世界林业研究, 2021, 34(4): 26-32.
ZHANG M M, WANG J H, LU N, MA W J, WANG N, WU X M. Research progress and application of whole genome selection in forest tree breeding. World Forestry Research, 2021, 34(4): 26-32. (in Chinese)
[14] KUMAR S, CHAGNÉ D, BINK M C A M, VOLZ R K, WHITWORTH C, CARLISLE C. Genomic selection for fruit quality traits in apple (× domestica Borkh.). PLoS ONE, 2012, 7(5): e36674.
[15] MURANTY H, TROGGIO M, BEN SADOK I, AL RIFAÏ M, AUWERKERKEN A, BANCHI E, VELASCO R, STEVANATO P, VAN DE WEG W E, DI GUARDO M, KUMAR S, LAURENS F, BINK M C A M. Accuracy and responses of genomic selection on key traits in apple breeding. Horticulture Research, 2015, 2: 15060.
[16] BISCARINI F, NAZZICARI N, BINK M, ARÚS P, ARANZANA M J, VERDE I, MICALI S, PASCAL T, QUILOT-TURION B, LAMBERT P, DA SILVA LINGE C, PACHECO I, BASSI D, STELLA A, ROSSINI L. Genome-enabled predictions for fruit weight and quality from repeated records in European peach progenies. BMC Genomics, 2017, 18: 432.
[17] MINAMIKAWA M F, TAKADA N, TERAKAMI S, SAITO T, ONOGI A, KAJIYA-KANEGAE H, HAYASHI T, YAMAMOTO T, IWATA H. Genome-wide association study and genomic prediction using parental and breeding populations of Japanese pear (Nakai). Scientific Reports, 2018, 8: 11994.
[18] NSIBI M, GOUBLE B, BUREAU S, FLUTRE T, SAUVAGE C, AUDERGON J M, REGNARD J L. Adoption and optimization of genomic selection to sustain breeding for apricot fruit quality. G3: Genes Genomes Genetics, 2020, 10(12): 4513-4529.
[19] 王力荣, 朱更瑞. 桃种质资源描述规范和数据标准. 北京: 中国农业出版社, 2005: 54-76.
WANG L R, ZHU G R. Descriptors and Data Standard for Peach (L.). Beijing: China Agriculture Press, 2005: 54-76. (in Chinese)
[20] MURRAY M G, THOMPSON W F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Research, 1980, 8(19): 4321-4326.
[21] CHEN S F, ZHOU Y Q, CHEN Y R, GU J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 2018, 34(17): i884-i890.
[22] LI H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013: arXiv: 1303.3997. https://arxiv.org/abs/1303. 3997.
[23] MCKENNA A, HANNA M, BANKS E, SIVACHENKO A, CIBULSKIS K, KERNYTSKY A, GARIMELLA K, ALTSHULER D, GABRIEL S, DALY M, DEPRISTO M A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 2010, 20(9): 1297-1303.
[24] YANG J, LEE S H, GODDARD M E, VISSCHER P M. GCTA: A tool for genome-wide complex trait analysis. American Journal of Human Genetics, 2011, 88(1): 76-82.
[25] VERDE L, ABBOTT A G, SCALABRIN S, JUNG S, SHU S Q, MARRONI F, ZHEBENTYAYEVA T, DETTORI M T, GRIMWOOD J, CATTONARO F,. The high-quality draft genome of peach () identifies unique patterns of genetic diversity, domestication and genome evolution. Nature Genetics, 2013, 45(5): 487-494.
[26] CAO K, LI Y, DENG C H, GARDINER S E, ZHU G R, FANG W C, CHEN C W, WANG X W, WANG L R. Comparative population genomics identified genomic regions and candidate genes associated with fruit domestication traits in peach. Plant Biotechnology Journal, 2019, 17: 1954-1970.
[27] HERNANDEZ C O, WYATT L E, MAZOUREK M R. Genomic prediction and selection for fruit traits in winter squash. G3: Genes Genomes Genetics, 2020, 10(10): 3601-3610.
[28] HONG J P, RO N, LEE H Y, KIM G W, KWON J K, YAMAMOTO E, KANG B C. Genomic selection for prediction of fruit-related traits in pepper (spp.). Frontiers in Plant Science, 2020, 11: 570871.
[29] LI Y L, RUPERAO P, BATLEY J, EDWARDS D, KHAN T, COLMER T D, PANG J Y, SIDDIQUE K H M, SUTTON T. Investigating drought tolerance in chickpea using genome-wide association mapping and genomic selection based on whole-genome resequencing data. Frontiers in Plant Science, 2018, 9: 190.
[30] WIMMER V, LEHERMEIER C, ALBRECHT T, AUINGER H J, WANG Y, SCHÖN C C. Genome-wide prediction of traits with different genetic architecture through efficient variable selection. Genetics, 2013, 195(2): 573-587.
[31] CALUS M P L, VEERKAMP R F. Accuracy of multi-trait genomic selection using different methods. Genetics Selection Evolution: GSE, 2011, 43(1): 26.
[32] HEFFNER E L, JANNINK J L, IWATA H, SOUZA E, SORRELLS M E. Genomic selection accuracy for grain quality traits in biparental wheat populations. Crop Science, 2011, 51(6): 2597-2606.
[33] HEFFNER E L, JANNINK J L, SORRELLS M E. Genomic selection accuracy using multifamily prediction models in a wheat breeding program. The Plant Genome, 2011, 4(1): 65-75.
[34] CAO K, ZHOU Z K, WANG Q, GUO J, ZHAO P, ZHU G R, FANG W C, CHEN C W, WANG X W, WANG X L, TIAN Z X, WANG L R. Genome-wide association study of 12 agronomic traits in peach. Nature Communications, 2016, 7: 13246.
Genomic Selection for Fruit Weight and Soluble Solid Contents in Peach
CAO Ke, CHEN ChangWen, YANG XuanWen, BIE HangLing, WANG LiRong
Zhengzhou Fruit Research Institute, Chinese Academy of Agricultural Sciences, Zhengzhou 450009
【Background】Fruit weight and soluble solid content (SSC) are two important quantitative traits in peach which are of importance to breeders. However, performing early prediction using a single marker is challenging as the traits are controlled by multiple minor genes. Genomic selection, a novel genome-wide tool, has been applied in fruit crops and can potentially enhance the breeding efficiency of these quantitative traits. However, its effects in peach and influencing factors require further investigation.【Objective】Establish a whole-genome selection technology system for peach single fruit weight and SSC, and laid a methodological foundation for the establishment of efficient molecular breeding technology system for peach.【Method】The objectives of this study were to assess the accuracy of prediction of peach fruit weight and SSC in natural and hybrid populations using genomic selection. Here, a training population of 520 individuals was selected. Using genotypic data for 48 398 single nucleotide polymorphisms (SNPs) obtained from the resequencing results of the above training population, a total of 11 genome-wide prediction models were built to select the optimum model for fruit weight and SSC. Subsequently, the genomic breeding values of a small natural population of 56 individuals and 29 hybrid populations comprising a total of 1 145 seedlings were calculated.【Result】The average sequencing data of each variety of the three groups was 1.95-3.52 Gb, and the sequencing depth was 5.29-10.79×. The sequencing data of the training natural population was aligned with the reference genome, and a total of 5 065 726 SNPs were obtained. After removing the SNPs with a high missing rate (>20%) and minor allele frequency of <0.05, a total of 48 398 SNPs on the genome were randomly selected for constructing whole-genome selection models for the training population. The models with the highest prediction accuracy for fruit weight and SSC were BayesA and randomforest, respectively. Using the above two models, it was found that the goodness of fit between the predicted breeding values and observed phenotype of fruit weight was 0.4767-0.6141, which was higher than that of SSC (0.3220-0.4329) in the natural populations. In hybrid populations, the prediction accuracy of fruit weight was 0.2319-0.4870, which was also higher than that of SSC (0.0200-0.2793). The results also showed that the prediction model constructed by training natural populations was more accurate in predicting natural populations than hybrid populations. Taking fruit weight as an example, it was also found that only 17.78% of the seedlings needed to be retained by genomic selection when targeting large fruit. Genomic selection was significantly more efficient than single and double marker selection. Furthermore, the effects of population dispersion, heritability and population structure on prediction accuracy are also discussed. The results indicated that prediction accuracy may vary and be affected by a combination of several factors.【Conclusion】In this study, a suitable genomic selection model for peach fruit weight and SSC was screened, and it was confirmed that the prediction efficiency of genomic selection was significantly higher than that of single marker selection. The results indicated the potential of genomic prediction in accelerating breeding progress of these two quantitative traits in peach.
peach; fruit weight; soluble solid contents; genomic selection; early prediction

10.3864/j.issn.0578-1752.2023.05.011
2022-04-28;
2022-09-09
中国农业科学院科技创新工程专项(CAAS-ASTIP-2020-ZFRI)
曹珂,Tel:13673618358;E-mail:wyandck@126.com。通信作者王力荣,Tel:13700883956;E-mail:wanglirong@caas.cn
(责任编辑 赵伶俐)
猜你喜欢
现代农业科技(2022年18期)2022-09-27
东北农业大学学报(2022年7期)2022-09-23
河北果树(2020年2期)2020-05-25
农家之友(2018年7期)2018-07-19
现代园艺(2017年21期)2018-01-03
中央民族大学学报(自然科学版)(2016年3期)2016-06-27
广西林业科学(2016年2期)2016-03-20
中国康复理论与实践(2015年10期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
