
Synthesis of allenyl-B(MIDA) via hydrazination/fragmentation reaction of B(MIDA)-propargylic alcohol
2023-03-14 06:52JiashengQianZhiHaoChenYuanLiuYinLiQingjiangLiShiLiangHuangHonggenWang
Chinese Chemical Letters 2023年1期
Jiasheng Qian,Zhi-Hao Chen,Yuan Liu,Yin Li,Qingjiang Li,Shi-Liang Huang,Honggen Wang
Guangdong Key Laboratory of Chiral Molecule and Drug Discovery,School of Pharmaceutical Sciences,Sun Yat-Sen University,Guangzhou 510006,China
Keywords:Allenyl boronate Mitsunobu reaction Mida boronates Hydrazination Fragmentation Chirality transfer
ABSTRACT Allenylboronates represent a very intriguing class of organoborons but are challenging to synthesis.In addition,these compounds are typically unstable,rendering the separation difficult.We report herein a practical and concise route to a new class of stable,easy-separable allenyl B(MIDA) via a hydrazination/fragmentation of B(MIDA)-propargylic alcohols.The synthesis of optically active allenyl B(MIDA) was also achieved.Interesting reactivity of the resulting product was observed.
Allenylboronate,with a boryl functionality directly attached to a cumulativeπ-system,have found intriguing applications in organic synthesis for the preparation of stereo- and regio-defined allenes or propargylic compounds [1–5].Nevertheless,the synthesis of allenylboronates has been challenging,although plenty of methods to access allenes are known [6].Only a handful of methodologies based on the final-stage installation of boryl moiety have been developed.For example,the reaction of reactive allenylmetallic reagents such as allenyl lithiums or allenylmagnesium bromides with trialkyl borate represent a straightforward synthesis of allenylboronates,but a strict low temperature is required and poor functional group tolerance is typically found (Scheme 1a)[2,4,5,7].The transition-metal-catalyzed hydroboration of enynes[8–12] or boryl substitution of propargylic compounds [13–18] offers an alternative access to multiple substituted allenylboronates(Scheme 1b).Still,these reactions hardly tolerate terminal alkynes(which readily undergo Glaser homocoupling),making them less suitable for 1,3-disubstituted allenylboronates synthesis.Of note,Studer group successfully realized a deoxygenative radical borylation of terminal propargylic alcohol derivatives.1,3,3-Trisubstituted allenylboronates were formed in moderate yields (Scheme 1c) [19].Very recently,Hoveyda reported a new method for the preparation of 1,3-allenylboronatesviacatalytic cross-metathesis of allenes.Excess amount of allenyl-Bpin was used with only moderate effi-ciency being observed (Scheme 1d) [20].
In addition to the shortage of synthetic methodology,the instability of allenylboronates brings about another challenge for their practical preparation.The so far reported allenylboronates (Bcat,Bpin,Bdan,etc.) are all unstable upon silica gel chromatography and decompose readily over long-term storage.
The emergence of protected tetracoordinate organoboron derivatives,such asN-methyliminodiacetyl (MIDA) boronates,as boronic acid surrogates has opened up new opportunities to potentially address both the stability and reaction efficiency of organoborons [21].Recent developments of B(MIDA) derivedα-boryl heteroaromatics/aldehydes/carboxylic acids andβdifluoroalkyl boronates have further highlighted the prominent effect of sp3boron [22–30].To tackle the nauseous instability of allenylboronates,we reasoned a protecting group strategy on boron might provide a viable solution.Nevertheless,although different types of organo B(MIDA) including alkenyl B(MIDA) [31],alkyl B(MIDA) [32–34],aryl B(MIDA) [35,36] and alkynyl B(MIDA) [37,38]were well studied,the corresponding allenyl B(MIDA) is thus for not known.In addition,unlike previous protocols featuring the final-stage installation of the boryl moiety,we anticipated the remarkable stability of organo B(MIDA) may offer it a chance for a late-stage modification [39,40],namely a late-stage construction of the allenyl moiety.
Herein,we describe a new method for the synthesis of 1,3-disubstituted and 1,3,3-trisubstituted allenyl B(MIDA) from easily accessible B(MIDA)-propargylic alcoholsviaa one-pot hydrazination/fragmentation sequence [41].A wide range of allenyl B(MIDA)s,which are typically white solid and stable towards silica gel chromatography,were prepared in good efficiency.The stereospecificity of this protocol also allowed the synthesis of optically active allenyl B(MIDA) from optically active propargylic alcohols,which are widely available.Initial studies showed that,the resulting sp3-B allenyl B(MIDA) have intriguing reactivities,distinct from their sp2-B congeners.
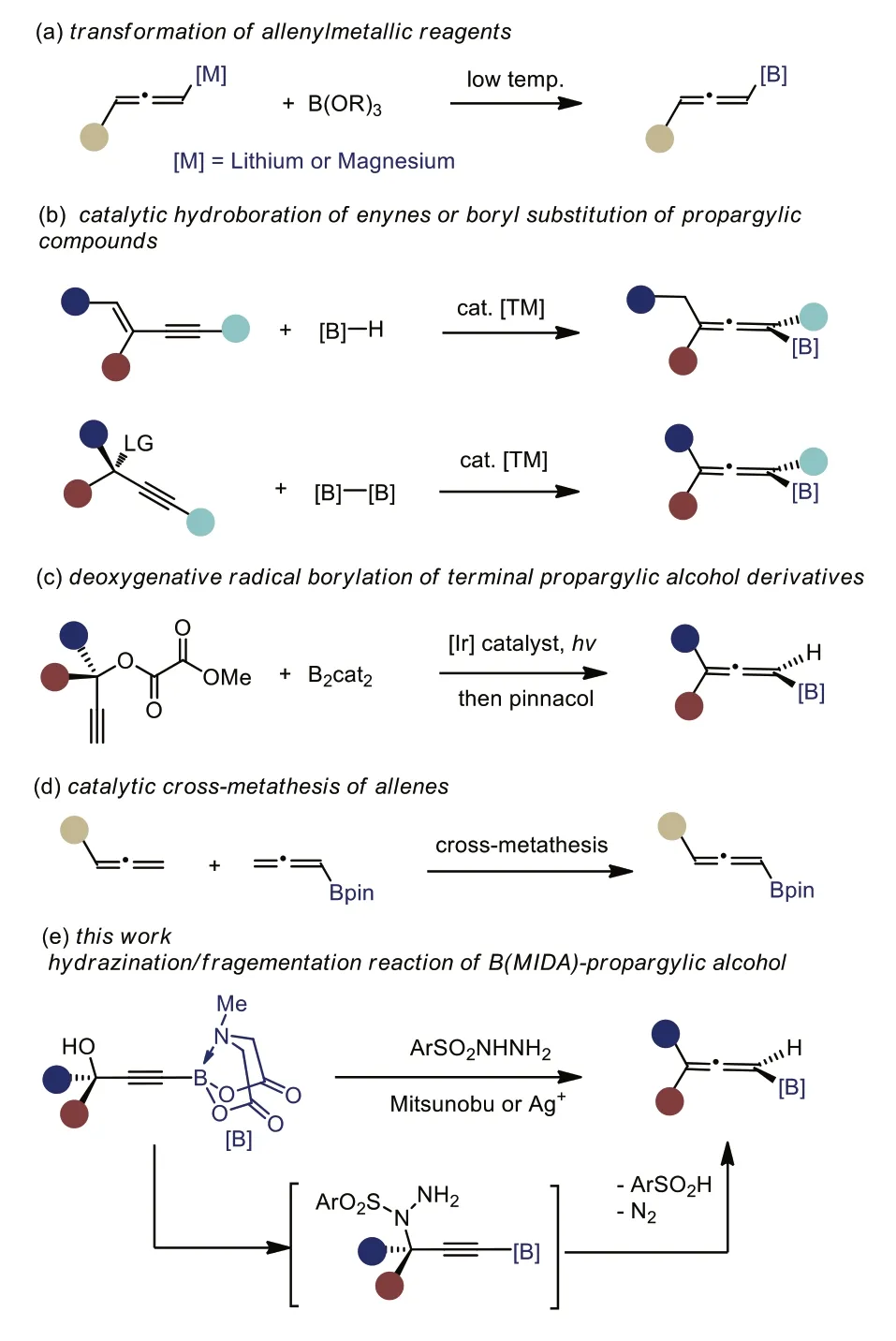
Scheme 1.Strategies for the synthesis of allenylboronate.
We commenced our study with B(MIDA)-propargylic alcohol 1a as a model substrate.By following Myers’ conditions comprising of PPh3(1.3 equiv.),DEAD (1.3 equiv.),NBSH (onitrobenzenesulfonylhydrazine,1.3 equiv.) in dry THF at -15 °C to room temperature [41],the desired product was isolatedviasilica gel chromatography in 37% yield as a white solid (entry 1,Table 1).While MeCN provided a slightly lower 30% yield (entry 2),other nonpolar solvents such as DCM and toluene turned out to be completely ineffective (entries 3 and 4).To improve the yield,efforts were then paid on the nature of Mitsunobu reagents and arenesulfonylhydrazine.Unfortunately,changing PPh3to PnBu3(entry 5)or DEAD to DIAD (entry 6) resulted in significantly inferior results.However,the replacement of NBSH with IPNBSH [42] was found to be also effective but gave a lower yield (entry 7).Unexpectedly,a profound yield improvement was observed by simply increasing the loading of Mitsunobu reagents (entries 8 and 9).Thus,with 2.0 equiv.of PPh3and DEAD,a good yield of 73% was obtained(entry 10).However,increasing the loading of NBSH gave no beneficial result (entry 11).The reaction was rather sensitive to the reaction temperature.Changing the reaction temperature to room temperature or 60 °C gave no desired product at all (entries 12and 13).And finally,the BF3K analogue of 1a,as another type of stable tetracoordinate organoboron,decomposed completely under the optimized reaction conditions (Eq.1),indicting the unique role of B(MIDA) in this reaction.We also attempted to synthesis the corresponding Bpin- or Bdan- propargylic alcohol,but failed.
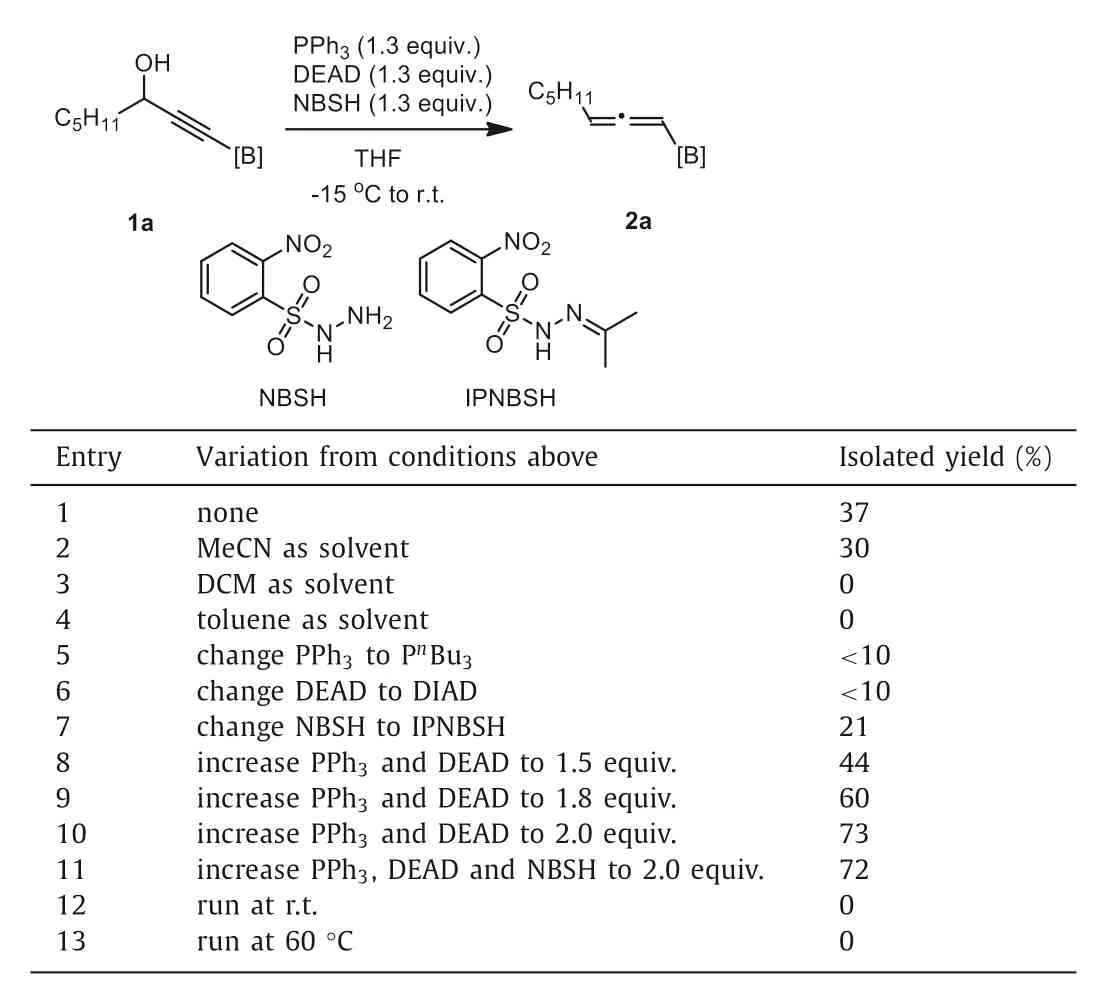
Table 1 Optimization of reaction conditionsa.
With the optimized reaction conditions in hand (entry 10,Table 1),we set out to investigate the scope and limitation of the method.As shown in Scheme 2,a series of primary (1a-1d) and secondary (1e-1j) alkyl substituents at the propargylic position were well tolerated,giving the corresponding 1,3-disubstituted allenyl B(MIDA)s in moderate to good yields.Phenyl (1k-1m) and furyl (1n) groups were compatible as well.Thecis-double bonds residing at the alkyl chain remained untouched (1o-1p).And some commonly encountered functional groups such as halides (1q-1r),ether (1s),benzyloxy (1t) and ester (1u) survived,demonstrating the mildness of this protocol.The moderate yields for some cases are due to the undesired amination with the amino group of dicarbethoxyhydrazine,the products of which are reluctant to undergo fragmentation reaction (see Supporting information for details).
Tertiary alcohols were typically not viable substrates in Mitsunobu reactions [43].Indeed,1v bearing two phenyl groups gave no expected allene product using the standard conditions (Scheme 3).We then resorted to a Lewis-acid catalyzed deoxygenation strategy [44].As shown in Scheme 3,with AgOTf as catalyst in the presence of NBSH,the desired 1,3,3-trisubstituted allenyl B(MIDA)2v was produced in 36% yield,along with 47% of unselective amination product 2v’as side product.Following this procedure,1w-1x bearing one phenyl group and one alkyl group gave 2w-2x in moderate yield.And the reaction ofp-tolyl substituted substrate gave 74% yield of 2y product.Unfortunately,2z and 2a were not formed in this sliver-catalytic reaction,with most the substrates being recovered.
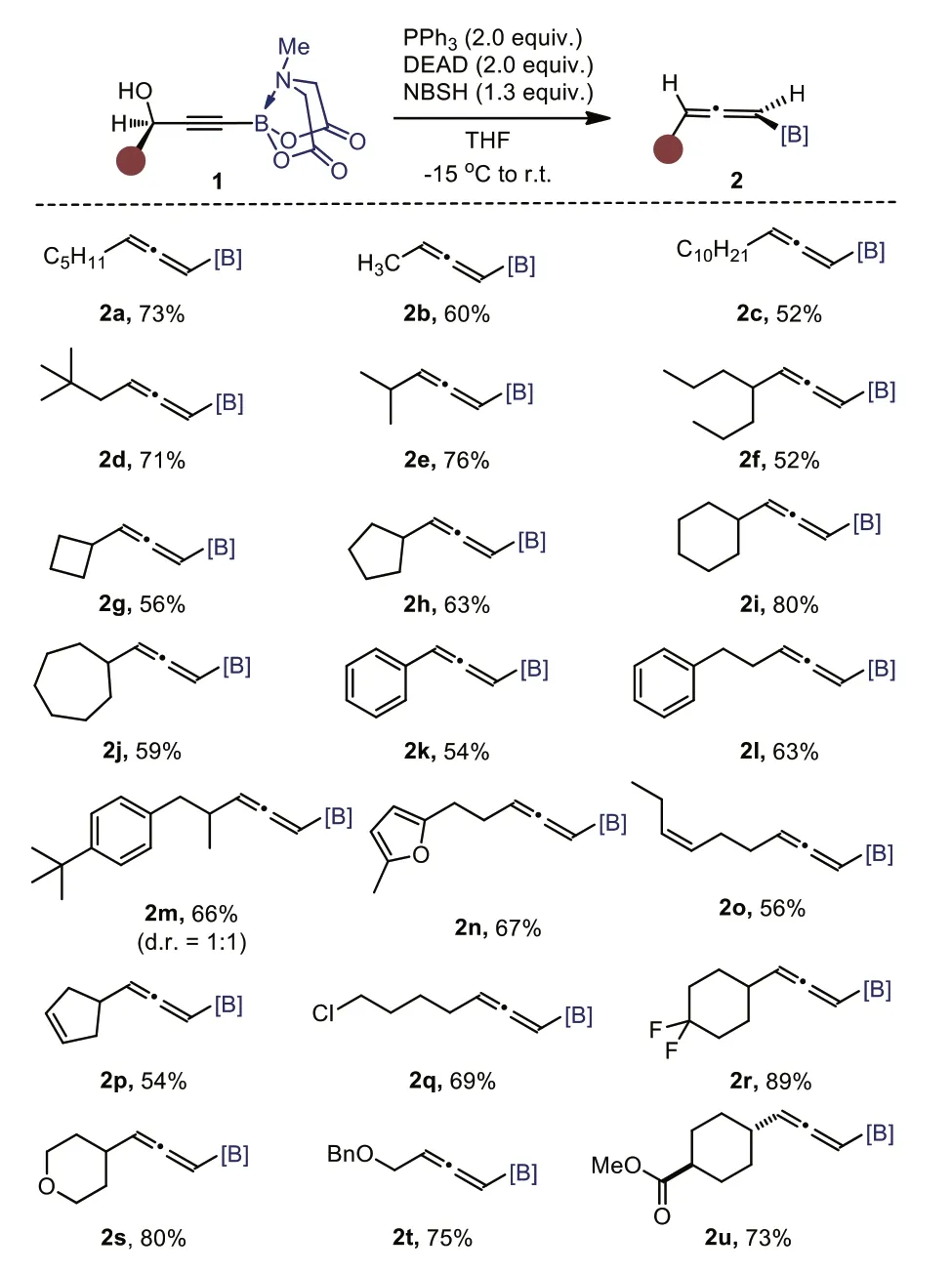
Scheme 2.Synthesis of 1,3-disubstituted allenyl BMIDA.
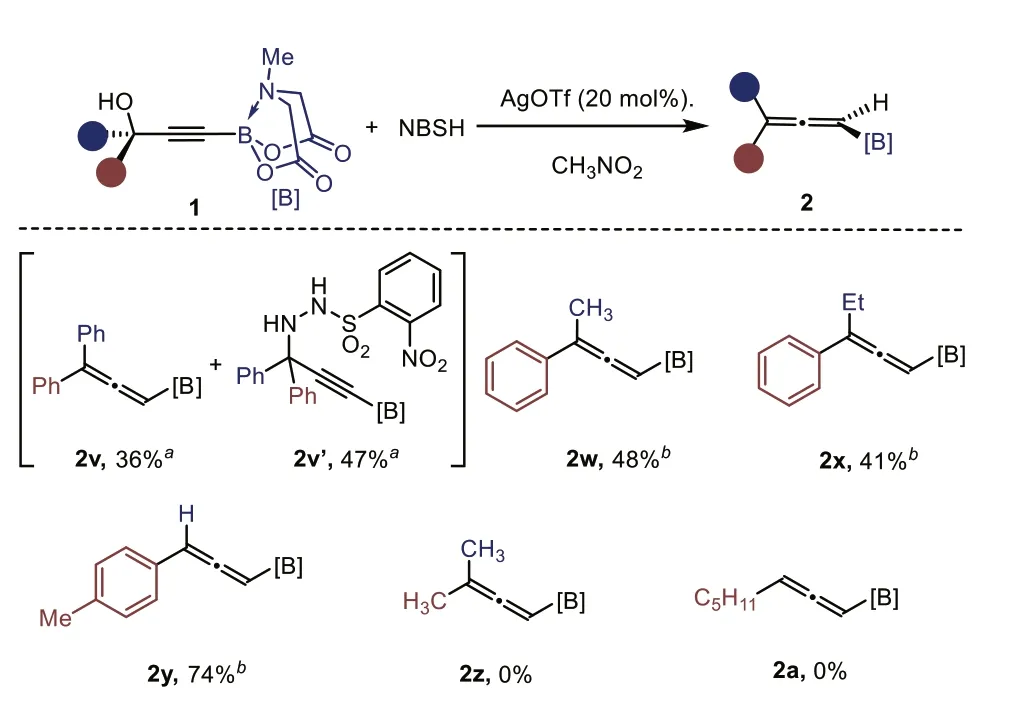
Scheme 3.Synthesis of allenyl BMIDA via a sliver-catalytic reaction with NBSH. a 35 °C,3 h. b 60 °C,1 h.
To showcase the practicality of this protocol,a gram-scale synthesis was conducted (Scheme 4a).Thus,the starting 1a was preparedviaa borylation of terminal propargylic alcohol SM1,which is commercially available,and follow-up ligand exchange in overall 46% yield without isolating the intermediate.Thereafter,the standard Mitsunobu reaction/fragmentation underwent smoothly to provide 2a in 79% yield (vs.73% in small scale),as isolated by normal silica gel chromatography without special precautions.It should be noted that 2a was stable upon storage in refrigerator at 0 °C for months.The stereospecificity of both the Mitsunobu reaction and fragmentation allowed a facile synthesis of optically active allenyl B(MIDA).For example,starting from enantioenriched B(MIDA)-propargyl alcohol (*)1k,readily prepared from CBS reduction of ynone in 70% yield,(*)2k was obtained in >99%eeunder standard reaction conditions (Scheme 4b).
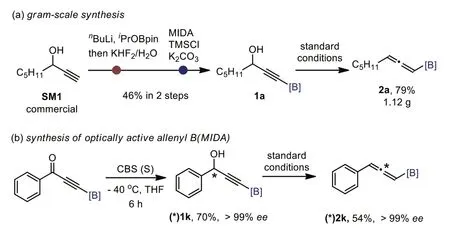
Scheme 4.Synthesis applications.
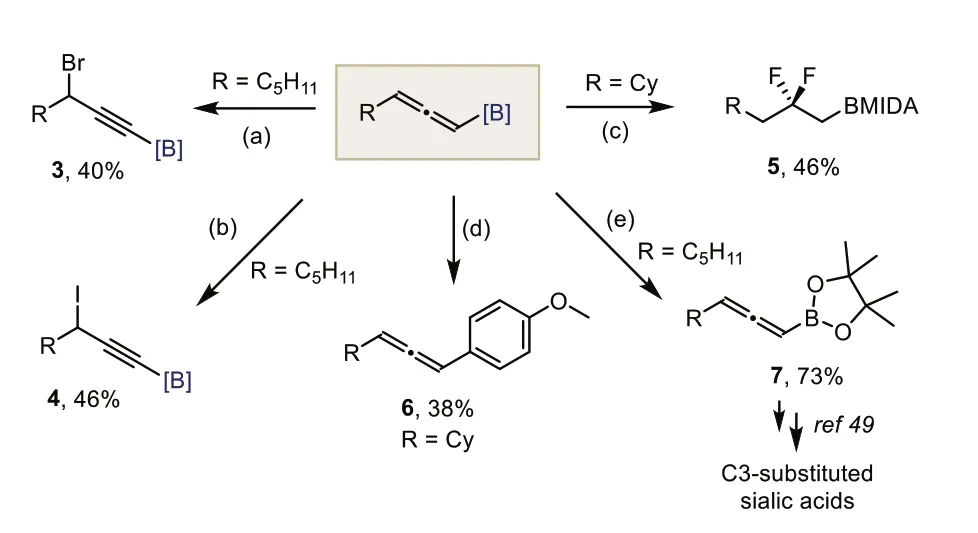
Scheme 5.Derivatizations of allenyl B(MIDA)s.Reaction conditions: (a) 2a(0.2 mmol),NBS (2.0 equiv.),acetone/H2O,23 °C,1 h; (b) 2a (0.1 mmol),DIH (1.0 equiv.) DCM/H2O,23 °C,5 min; (c) 2i (0.1 mmol),Py-HF (40.0 equiv.),DCM (0.3 mL),23 °C,30 min; (d) 2i (0.1 mmol),1-iodo-4-methoxybenzene (1.5 equiv.),Pd(OAc)2 (5 mol%),SPhos (10 mol%),K3PO4 (7.5 equiv.),THF/H2O,60 °C,24 h; (e) 1a (0.2 mmol),NaHCO3 (5.0 equiv.),pinnacol (5.0 equiv.),MeOH,50 °C,2 h.
Owing to the two orthogonal C=C double bonds,allenes have exhibited high reactivity and chemical diversity in modern synthetic transformations [45–47].To assess the synthetic utility of the resulting allenyl B(MIDA),we then tested the reactivity of allenyl B(MIDA) in several transformations (Scheme 5).The reaction of 2a with NBS in acetone/H2O gave a propargylic brominated product 3 in 40% yield,with the B(MIDA) moiety being retained.Likewise,iodide 4 was formed in 47% yield by treating 2a with DIH.Interestingly,treating 2i with Py-HF delivered a valuableβdifluoroalkylboron 5viaregioselective bis-hydrofluorination.Transformations of the C–B bond were also possible.A Suzuki–Miyaura cross-coupling with 1-iodo-4-methoxybenzene under unoptimized slow-release conditions afforded allene 6 in 38% yield.And finally,ligand exchange of 2a smoothly furnished the corresponding allenyl pinacol boronic ester 7,which was a known building block for multiple utilities,including the synthesis of C3-substituted Sialic acids (see Supporting information for details) [48,49].
In summary,a new synthesis of allenylboron from preborylated propargylic alcoholsviaa one-pot Mitsunobu Reaction/fragmentation has been developed.For aromatic substrates,a silver-catalytic reaction with NBSH offered an alternative method.The presence of B(MIDA) moiety conferred not only high stability,but also intriguing reactivity to the resulting products,as demonstrated by several boryl-retentive transformations.The Mitsunobu Reaction/fragmentation process was found to be stereospecific.Given the wide availability of methods to access optically active propargylic alcohols,the developed protocol should also find applications in the synthesis of diverse optically active allenylborons.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos.22022114 and 21971261),the Guangdong Basic and Applied Basic Research Foundation (No.2020A1515010624),the Fundamental Research Funds for the Central Universities (No.20ykzd12) and the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (No.2017BT01Y093).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.04.077.
 Chinese Chemical Letters2023年1期
Chinese Chemical Letters2023年1期
- Chinese Chemical Letters的其它文章
- Diabetic wound healing activated by supramolecular cascade reaction
- MBenes: Two-dimensional transition-metal borides with ordered metal vacancies
- Wet-adhesive materials of oral and maxillofacial region: From design to application
- Diverse catalytic systems for nitrogen-heterocycle formation from O-acyl ketoximes
- Fluorine-containing drugs approved by the FDA in 2021
- The development and application of dual-comb spectroscopy in analytical chemistry