
I2/PhI(OAc)2-assisted oxidative C–H amination protocols toward metal-free pragmatic synthesis of pyrrolo[2,3-b]indoles
2023-03-14 06:52YupingXiongFeihuDuLinghuiZengZhiyunChen
Chinese Chemical Letters 2023年1期
Yuping Xiong,Feihu Du,Linghui Zeng,*,Zhiyun Chen,,*
a College of Chemistry & Chemical Engineering,Jiangxi Normal University,Nanchang 330022,China
b School of Medicine,Zhejiang University City College,Hangzhou 310015,China
Keywords:C–H activation C–H amination Indole Oxidative cyclization aza-Heterocycle
ABSTRACT We report herein an I2/PhI(OAc)2 catalytic system for the pragmatic construction of C–N bonds through C–H/N–H oxidative coupling protocol.Divergent pyrrolo[2,3-b]indoles were efficiently prepared via I2-catalyzed intramolecular C–H amination reactions from (E/Z)-2-indolylenamines under metal-free conditions.Various functional groups are tolerated under mild reaction conditions and the resulting pyrrolo[2,3-b]indoles were obtained with mostly good to excellent yields.It was interesting to observe that both the (E)- and (Z)-isomers of the starting materials were efficiently transformed into the targeted product.The I+-mediated catalytic cycle was proposed based on mechanistic studies for this reaction.
Indoles are among the most privileged medicinal scaffolds that can be frequently found in natural products,as well as in various pharmaceutical candidates,dyes,and agrochemicals [1–3].In particular,structurally diversified indolyl fused pyrrolines are biologically valuable natural alkaloids (Fig.1).For instance,pyrrolo[2,3-b]indole derivatives such as pyrroindomycins A and B[4] were isolated fromStreptomyces rugosporusin 1994 [5,6].Recently,families of these compounds were found to behave potent antibiotic activities against bacterial pathogens,and in vitro activity against Gram-positive bacteria [7].The 5,6-dihydroindolo[2,3-b]indoles also demonstrated excellent activities as Sirtuins inhibitors [8,9],or as growth inhibitors of Bacillus subtilis [10].Due to their prominent pharmaceutical activities,pyrrolo[2,3-b]indole frameworks have always been the superior choice in medicinal studies,and their synthetic methodologies have also received extensive attention.
Recently,transition-metal-catalyzed annulations leading to fused aza-heteroaromatics have attracted particular attention.To date,the synthesis of indole-based fused polyheterocycles that were catalyzed by varied transitionmetals,such as Pd [8,11,12],Au [13],Cu/[O] [14],Fe [15] or Zn/NBS [16],were reported.Nevertheless,formidable limitations with the conventional cyclizations or cycloadditions still exist,mainly due to their reliance on specific active metal catalysts.As a supplement,metal-free protocols can offer quick but low-cost access to diverse molecular structures.Indeed,metal-free reactions have shown great potential for rapid production of complex aza-heterocyclic molecules in a single synthetic operation,which is of exceeding importance in the large-scale preparation of medicinal-related substances by avoiding metal pollution [17–20].In 1996,an interesting heatpromoted stepwise Barton-Zard reaction was developed to deliver a pyrrolo[2,3-b]indole ring system [21].Besides,stoichiometric amounts of hypervalent iodine reagent [22,23] or NIS [24] mediated amination of indole frameworks have also shown potential towardN-arylated andN-alkylated fused aza-heterocycles.These methods allowed the rapid construction of the indole skeletons by connecting N atoms on the side chain of the phenyl moieties.However,compared with the noticeable C3-functionalization,the C2-amination of indole is less explored.Some limited reports are available for intramolecular C2-amination of indole scaffolds.In this regard,Wang disclosed I2-mediated oxidative coupling of intramolecular C–N bond formations to afford indole derivatives.Wang group disclosed an iodine-catalyzed intramolecular amination of tryptophan esters for the practical synthesis of pyrrolo[2,3-b]indoles (Scheme 1a) [25].Sekar and co-workers developed I2-mediated intramolecular C2 sulfonamidative cyclization of indoles to give indole fused tetracyclic compounds (Scheme 1b) [26].While these methods should shed light to the synthesis of pyrrolo[2,3-b]indole derivatives,step-wise preparation of the starting materials,and/or pre-functionalization of an electronwithdrawing substituent at the certain position of indolyl precursors are often necessary.
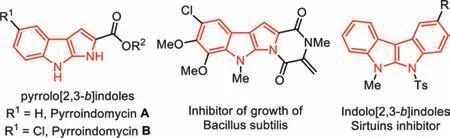
Fig.1.Selected biologically active pyrrolo[2,3-b]indole derivatives.
Our group previously reported a 1,3-insertion reaction of a Rhcarbenoid into the C(sp2)-H bond of simple indoles [27],and subsequently sequential C–H oxidation to selectively produce (E/Z)-2-indolylenamides or (Z)-3-arylidene-2-oxindole imides [28].As part of our interest in the fused polyheterocyclic alkaloids [29–33],we envisioned that an I2-catalyzed C2-amination of C–H/N–H oxidative coupling should offer a pragmatic synthesis of structurally divergent pyrrolo[2,3-b]indoles (Scheme 1c).Herein,we report a highly efficient metal-free protocol to chemo- and regioselectively afford pyrrolo[2,3-b]indoles under very gentle conditions.
We sought to employ (E/Z)-mixture of 2-indolylenamide 1a as a model substrate to investigate this C2-aminative cyclization reaction (Table 1).The optimization commenced with 1a in the presence of iodine (5 mol%),oxidant [O],and Na2HPO4(3.5 equiv.) in toluene (2.0 mL) at 80°C.Indeed,the desired product 2a could be formed when Cu(OAc)2(1.05 equiv.) was selected as the oxidant,albeit in quite a low yield (entry 1).The configuration of compound 2a was determined by single-crystal X-ray diffraction analysis (CCDC: 2122559 (2a) in Supporting information for details).Product 2a was formed in 29% yield whentBuOOH was added instead of Cu(OAc)2(entry 2).To our delight,improved yields were observed when BQ or TBHP was added,and a 62% yield of product 2a was obtained in the presence of TBHP (entries 3 and 4).However,decreased efficiency was observed when I2loading was improved to 10 mol%,whereas a dramatically sluggish reaction was observed by reducing the amount of iodine to 2 mol%.These results indicated the crucial role of iodine species in this amidation protocol (entries 5 and 6).The reaction was optimistic for PhI(OAc)2,which could afford the desired product 2a in 68% yield(entries 7 and 8).Further screening of the reaction conditions revealed LiOH was a suitable base (entries 9–12).Pleasingly,we found that reducing the amount of LiOH to 1.2 equiv.did not restrain the transformation (entry 13).Among the solvent evaluation,the full transformation was observed in 1,4-dioxane in 8 h,and as a result,83% yield of 2a was produced (entries 14–16).Interestingly,a dilute 1,4-dioxane solution and reduced temperature were proved optimal for this reaction,which led to 96% yield of 2a when 3.0 mL of 1,4-dioxane was utilized at 50 °C (entry 17).Lowering the temperature to room temperature resulted in inferior transformation and only a modest yield of 2a was obtained (entry 18).Interestingly,the possible side product 2a_1,which was presumably formed from (E)-1a,was not observed in this catalytic system.And the fact that the almost quantitative yield of 2a demonstrated that this product should be originated from both (Z)-1a and (E)-1a,and thus mechanistically a common imine (C=NTs) intermediate should be the key species from the (E/Z)-1a substrates.
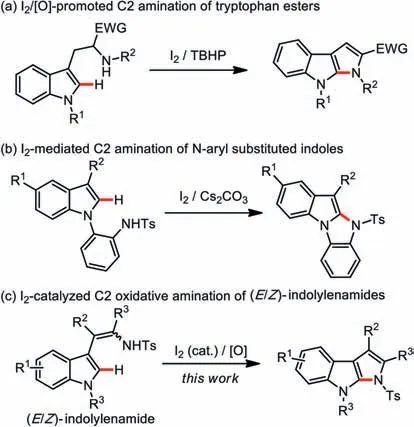
Scheme 1.I2-mediated intramolecular C2-amination of 2-indolylenamides.
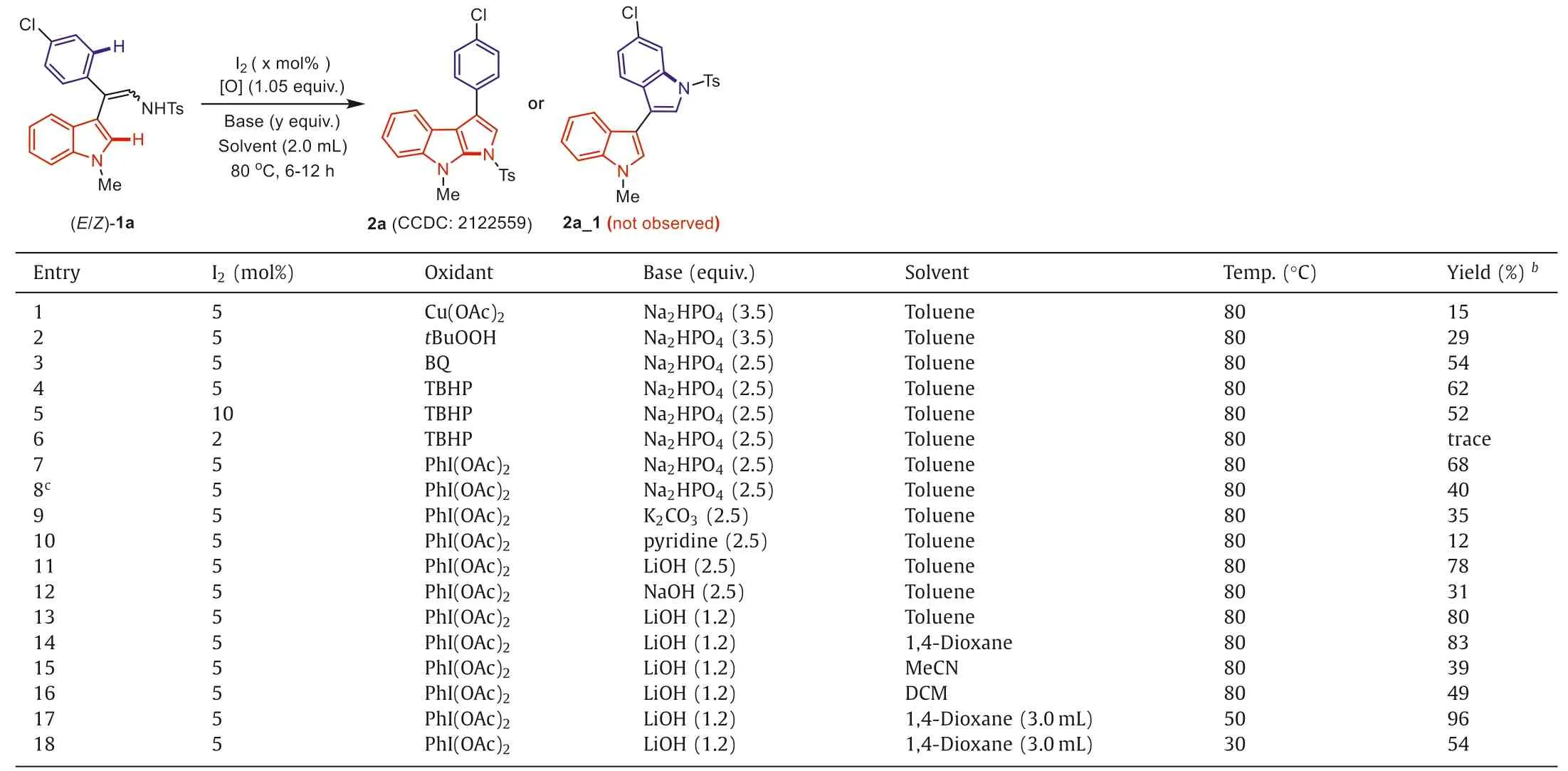
Table 1 Optimization of the reaction conditions.a
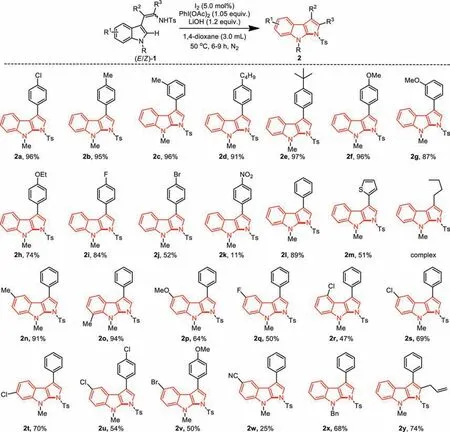
Scheme 2.Pragmatic synthesis of pyrrolo[2,3-b]indoles.Reaction conditions: (E/Z)-1 (0.20 mmol),I2 (5 mol%),PhI(OAc)2 (1.05 equiv.),and LiOH (1.2 equiv.) in 3.0 mL of 1,4-dioxane at 80 °C,5–8 h unless otherwise noted.Isolated yield based on (E/Z)-1.
With the optimized reaction conditions in hand (Table 1,entry 17),we next explored the substrate scope and the results are presented in Scheme 2.Different aromatic substituents at R2position of (E/Z)-2-indolylenamides 1 were firstly studied.The results indicated that varied functional groups with electron-donating properties were well tolerated.For example,aryl groups at R2could be attached with Me (2b and 2c),MeO (2f and 2g),EtO (2h),nBu(2d),andtBu (2e),delivering the corresponding products in good to excellent yields.Other than chloride (2a),the synthetically useful halide groups,such as F (2i) and Br (2j) at the aryl moiety of R2also worked smoothly to afford the expected products in 84% and 52% yields,respectively.Note that the strong electron-withdrawing nitro group at the aryl group of R2was less reactive,providing the desired product 2k in a hamper yield.Pleasingly,the heteroaryl 3-thienyl group was compatible for this C2-amidation of indole scaffold,delivering the desired product 2m in 51% yield.Unfortunately,the alkyl-substitution located at the R2position of 1 was intolerable under this I2-catalyzed oxidative condition,because the enamide 1 with a butyl-group at R2could be easily decomposed and no desired product could be detected.
For the substituents R1that were attached on to the benzenoid ring,results showed different functional groups with electrondonating or electron-withdrawing properties were all compatible (Scheme 2).Methyl groups which located at the 5- or 7-positions of indolyl benzoid moiety worked well to produce the desired products 2n and 2o in nice yields.Whereas 64% yield of 2p was obtained for the cyclization of 5-MeO substituted substrate.In addition,2-indolylenamides 1,which bearing an electronwithdrawing halo-substitution such as fluoro (2q),chloro (2r,2s,2t and 2y),and bromo (2v),were all well-tolerated without diffi-culty,regardless of their locations at the indole phenyl moieties.It was worth noting that the strong electron-withdrawing group like cyanide (2w) was also admissible tolerant,highlighting the possible subsequent divergent conversions of the products.In the case of theN-protecting group in the indolyl enamide,the benzyl group was found to be an optimal choice.The Bn-protect product 2x could be isolated from a Bn-substituted 2-indolylenamide precursor 1 in 68% yield under the standard conditions.Gratifyingly,by modifying the substituted enamide as a substrate,we were able to obtain the fully substituted pyrrole product 2y in 74% yield under the I2-catalyzed optimal conditions.
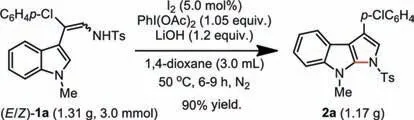
Scheme 3.Gram-scale synthesis of product 2a.
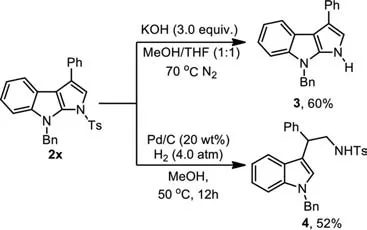
Scheme 4.Deprotection of the N-Ts.
The utility of this C–H/N–H coupling reaction can be demonstrated by a preparative gram-scale reaction,as the product 2a can be isolated in 90% yield on the 3.0 mmol scale of (E/Z)-1a(Scheme 3).
Deprotection of the tosyl group from compound 2x using KOH(3.0 equiv.) as a base [34] in MeOH/THF solvent was successful to obtain product 3 in a synthetically useful yield (60%).The attempts to remove the benzyl group in 2x under Pd/C and low-pressure H2conditions were failed.However,a ring-opening product 4 was isolated under 4.0 atm of H2reduction conditions in MeOH at 50°C(Scheme 4).
Mechanistic studies were carried out to understand the mechanism of this reaction (Table 2).When typical radical scavengers,such as TEMPO and BHT were added,the product 2a could be isolated in modest to good yields,suggesting that a free radical pathway should be unlikely involved in this indolyl C2 amination reaction (entry 1).LiOH worked as a base and was found to be necessary for this reaction (entry 2).With a catalytic amount ofI2,several different types of oxidants could participate in this reaction to deliver the desired product 2a (Table 1,entries 1–4).It was found that the stoichiometric amounts of I2could promote this cyclization protocol smoothly while in the absence of an oxidant (entry 3),however,no reaction occurred when PhI(OAc)2alone was added while in the absence of I2(entry 4).For the stoichiometric amounts of I2-promoted reactions of (E/Z)-1,we found that most of the reactions were achieved in humble yields,and inseparable mixture of the desired product with unknown side product were observed.These results clearly indicated that an I2–mediated catalytic cycle was involved in this transformation,and the oxidant should oxidize the molecular I2to offer a high reactive (hypo)iodites “I+” species in this iodine catalytic cycle.Indeed,“I+” species have been previously studied as powerful electrophiles which can react with C(sp2)-H substrates to give iodointermediates [35,36].
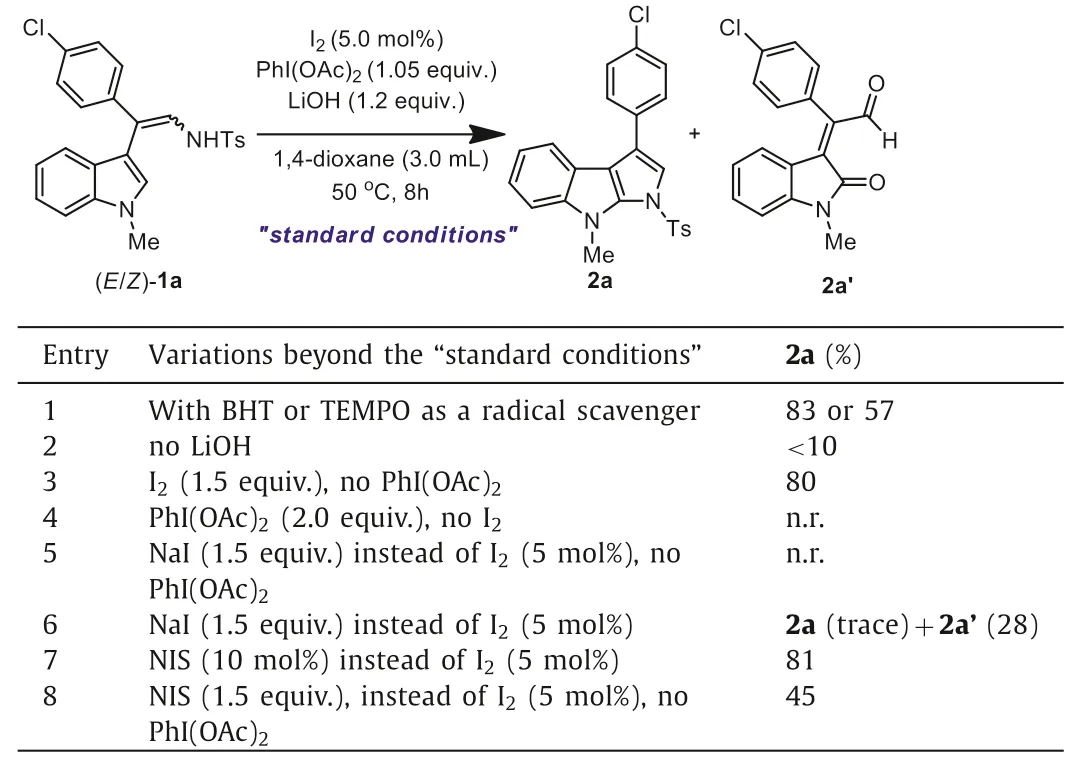
Table 2 Mechanistic studies.
To confirm the possible existence of I+,NaI (1.5 equiv.) was added instead of the catalytic amount of I2(5 mol%),we found that in this case,no reaction occurred when in the absence of oxidant (entry 5).Surprisingly,when NaI (1.5 equiv.) combined with 1.5 equiv.of PhI(OAc)2were added,the desired product 2a was only traced,but the hydrolysis side product 2a’was isolated in 28% yield (entry 6).The formation of aldehyde 2a’was observed in our previously Cp*IrIII-catalyzed sequential C–H oxidation of (E/Z)-indolyl-3-enamines 1 followed by a TsOH-promoted hydrolysis reaction [28].The observation of 2a’in the current I2-catalyzed reaction possibly indicated an indolyl C2-H oxidation to form the indolin-2-one intermediate be involved in this reaction.Further mechanistic studies using catalytic (entry 7) or stoichiometric (entry 8) amounts of NIS also strongly support the involvement of the I+-cation species in the I-cycle in this reaction.
Based on the abovementioned results,we propose a possible mechanistic rationalization as illustrated in Scheme 5.The reaction was initiated by the reaction of (E/Z)-1a with LiOH,to form a nitrogen-centered anion species B,which was followed by nucleophilic iodination reaction with molecular I2to give intermediate C upon release of I-anion.In the oxidant [O] promoted I-cycle,I-could be oxidized to I2to complete the catalytic cycle [37].In the following transformation,due to the electron-rich property of the indole,the conjugatedπ-system pushed an intramolecular heterolytic cleavage of the N-I bond in C to form ionic imine species D and its resonance imine isomer D’.The formation of imine (C=NTs) D or D’could explain why both of the stereoisomers (E)-1a and (Z)-1a all transformed into compound 2a.In addition,for the reaction of (E/Z)-1 with an electron-donating functional group at its R2position,such as a Me,tBu,and OMe group,the corresponding products 2e and 2f were obtained in almost quantitative yields,whereas for the reaction of (E/Z)-1 with a strong electron-withdrawing functional group,the corresponding products 2k and 2w,the yields of the expected products were only humble.These results should support the formation of carbocation D and resonance isomer D’.In the subsequent reactions,D’could be trapped by H2O to give the intermediate E.The following oxidation of E produced indolin-2-one F,which was followed by a hydrolysis reaction to deliver the aldehyde side product 2a’.Meanwhile,intermediate D’could also be trapped by I-to generate the 2-iodoindoline species G.The subsequent nucleophilic attack and cyclization reaction of G produced pyrroloindoline cation H,and finally,this species transformed into product 2a while with the assistance of LiOH.
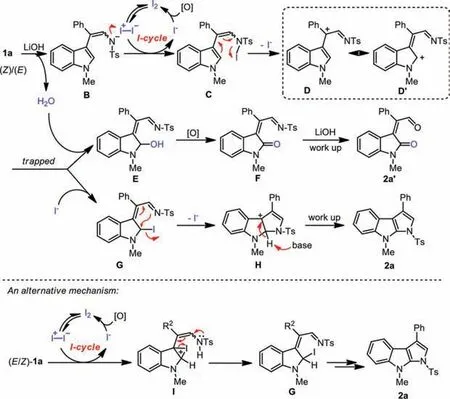
Scheme 5.Proposed mechanism.
An alternative mechanism was described as below in Scheme 5.In this mechanism,the indole moiety goes through electrophilic iodination I,and then ring-opening of the resultant iodonium salt gives the intermediate G.While this mechanism seemed reasonable,there’s a key point that this putative pathway cannot be clarified: since the electronic effect was not very obvious from this alternative mechanism,thus,the reason is not clear why substrate(E/Z)-1 with electron-donating groups,which are located at R2position,was much more reactive than those of substrates 1 with electron-withdrawing functional groups (2k and 2w).However,in the mechanistic rationalization,as illustrated above in Scheme 5,the stability of the carbocation D and D’could explain this phenomenon very well.
In summary,I2/PhI(OAc)2catalytic protocol was developed to promote the intramolecular oxidative C–H/N–H amination of(E/Z)-2-indolylenammides.The reaction worked smoothly under environmentally benign conditions and the desired pyrrolo[2,3-b]indoles were generated in mostly good to excellent yields under metal-free conditions.In light of the green and environmentally benign strategy that utilizing inexpensive I2as a catalyst to promote C–N bond formation sequences toward fused aza-heterocycles,this method should be potentially served as a pragmatic synthetic approach for the late-stage modification of pyrrolo[2,3-b]indoles,such as Pyrroindomycin analogues.Detailed mechanistic studies indicated an I+-mediated catalytic cycle was proposed in this reaction.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank the National Natural Science Foundation of China (No.21961015) and the Natural Science Foundation of Jiangxi Province(No.20202ACBL203005) for financial support.Y.Xiong acknowledges the support from the Jiangxi Provincial Education Department Graduate Student Innovation Fund (No.YC2021-S236).
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2022.05.083.
 Chinese Chemical Letters2023年1期
Chinese Chemical Letters2023年1期
- Chinese Chemical Letters的其它文章
- Diabetic wound healing activated by supramolecular cascade reaction
- MBenes: Two-dimensional transition-metal borides with ordered metal vacancies
- Wet-adhesive materials of oral and maxillofacial region: From design to application
- Diverse catalytic systems for nitrogen-heterocycle formation from O-acyl ketoximes
- Fluorine-containing drugs approved by the FDA in 2021
- The development and application of dual-comb spectroscopy in analytical chemistry