
家族性Holt-Oram综合征1例并文献复习
2023-03-08 16:38:28刘玉清
临床荟萃 2023年1期
刘玉清,程 佶
(天津市儿童医院/天津大学儿童医院 心脏中心,天津 300074)
Holt-Oram综合征(Holt-Oram Syndrome,HOS)是以心脏异常合并上肢骨骼畸形的常染色体显性遗传的单基因遗传病[1]。1960年Holt和Oram[2]报道过一个家族4代9例的患者,证明此症可具有鲜明的家族聚集性,但仍有散发的病例。本文将阐述由1例HOS患儿追溯到一个家族4代6例的病例。
1 临床资料
患儿系1个月男婴,主因“发现先天性心脏病5 d,发热9 h”于2021年7月9日就诊于我院。入院查体:营养发育可,意识清,反应可,呼吸稍促,无发绀。颜面部皮肤黄染,目测胆红素5 mg/dl。前囟平软,张力不高,大小约2.0 cm×2.0 cm,巩膜黄染,咽充血,双肺呼吸音粗,可及痰鸣音,心前区无隆起,未及震颤,心界不大,心音有力,心率135次/min,律齐,胸骨左缘第3~4肋间可及Ⅱ-Ⅲ/Ⅵ级收缩期杂音。腹软,肝肋下2 cm,质软边锐,脾肋下未及。双手拇指缺如,左上肢自然位腕部向桡侧屈曲,双下肢正常,四肢肌张力正常。神经系统查体未及异常。家族史:母亲患“房间隔缺损、左手拇指关节缺如”,外祖母患“房间隔缺损,左手拇指关节缺如”,曾外祖母患“左手拇指关节缺如”,舅姥爷有“漂浮指”、表舅有“漂浮指、房间隔缺损”。
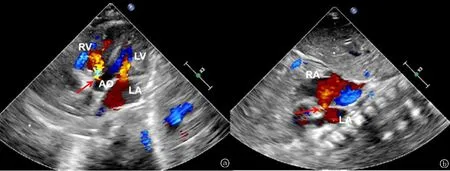
图1 患儿先天性心脏病的超声心动图表现 a.心尖五腔心切面可见膜周部室间隔缺损(↑);b.剑突下双房切面,可见房间隔缺损(↑)
患儿胸部电子计算机断层扫描示:双肺散在炎性实变,双肺纹理增重伴透过度不均,心影增大。心电图示窦性心律,Tv1直立,部分导联P波高尖,PR间期0.15 s,高于正常上限。超声心动图示各心腔内径均在正常范围内,房间隔中部可见直径约4 mm×4 mm回声失落,可见左向右分流血流,室间隔膜周部可见多处左向右分流血流,分流术最宽处约2 mm。各瓣口未见明显反流,未见心包积液及赘生物。提示房间隔缺损(中央型),室间隔缺损(膜周部、多发),见图1。手正位及尺桡骨正侧位片:双手第一掌骨及双拇指缺如,双侧桡骨较短,左侧尺桡骨近端融合,见图2。对其父母及患儿进行了全外显子组测序,检测到先证者12q24.21区域存在约0.6 Kb的片段杂合缺失,遗传自母亲。该缺失区域覆盖TBX5基因整个Exon9,可能引起HOS。
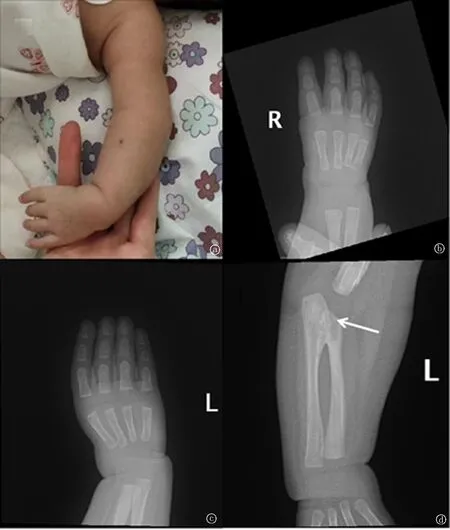
图2 患儿上肢畸形的临床表现及影像学异常 a.左手拇指缺如,左上肢自然位腕部向桡侧屈曲;b、c.右手及左手拇指缺如;d.左侧尺桡骨近端融合(↑)
2 讨 论
HOS的鲜明特征是先天性心脏病和上肢骨骼畸形,尤其是手部畸形,因此又被称作心-手综合征、心手症候群,发病率为1/100000活产儿[3],男女患病率无明显差异,虽特征鲜明,但不同患儿间的临床表现变异较大。
该综合征心脏异常的发生率为85%~95%,从无症状的传导系统异常到多种结构性心脏畸形均有报道,Newbury-Ecob等[4]研究中的55例HOS患者中,66%的患者发现了单纯性先天性心脏病,17.5%的患者发现了更复杂的先心病;其中家族性发病患者中39%仅表现为心电图异常,无先心病。Holt和Oram[2]曾报道的异常家族聚集性病例存在窦性心动过缓、PR间期延长和交界性逸搏, 此后有人报道还可出现右束支阻滞、窦房结功能障碍、心房颤动/心房扑动、阵发性室上性心动过速等心脏传导功能异常。该病常见的心脏畸形包括房间隔缺损、室间隔缺损、动脉导管未闭、心内膜垫缺损、肺静脉异位引流、法洛四联症等[5],其中以继发孔型房间隔缺损和室间隔缺损最为常见。Singh 等[6]报道称该病亦可出现血管异常,包括缺如或发育不良的外周动脉和静脉,如肾动脉和脑动脉畸形、外周上肢血管发育不全、左桡动脉发育不全、肺动脉高压。Misra等[7]报道1例HOS患者合并右上腔静脉缺如,残存左上腔静脉,术中麻醉因动脉和静脉置管不畅,行食道超声时发现血管异常,临时更改了体外循环的插管方式。本例患儿通过查体和超声心动图发现房间隔缺损及室间隔缺损同时出现,心电图提示PR间期延长。
HOS主要累及上肢骨骼,下肢畸形鲜有报道[8]。这可能是因为在胎儿生命中肢体和心脏的主要发育同时发生,上肢芽首先出现,并先于下肢芽完成生长,造成先天性缺陷优先累及上肢。上肢畸形程度轻重不等,双侧或单侧畸形,双侧畸形不对称,以桡侧多见,轻者可表现为手指弯曲变形、斜肩,重者出现海豹肢症等。上肢畸形中最常见的是拇指畸形,可表现为三指畸形、发育不良或完全缺如。在1项对98例拇指发育不良的受试者的研究中,16%的人患有HOS[9]。临床上可以通过体格检查和上肢X线摄片识别上肢畸形。本例患儿双侧拇指缺如,影像学提示双侧桡骨较短,左侧尺桡骨近端融合,为典型HOS综合征的上肢畸形。
HOS患儿可伴有其他先天性畸形:肺发育不全、心肌病、先天性巨结肠病、克罗恩病、泌尿系统畸形、隐睾症、Rokitansky-Kuster-Hauser综合征(未成熟或无阴道或子宫),还可伴发多次中风,近视,恶性肿瘤等。
对HOS患者的分子遗传学研究发现,TBX5基因变异是HOS的致病原因[10]。TBX5是转录因子基因家族的一员,其特征是高度保守的结合域称为T-box,它位于常染色体12q24.1,由10个外显子组成,对于上肢形成和心脏发育至关重要。通过动物模型显示出类似的缺陷,为研究TBX5在胚胎发育过程中的作用提供了一个有用的平台。研究证实在早期心脏发育过程中,TBX5似乎主要作为心肌细胞成熟相关基因的转录激活因子和分离形态学信号的上游。在后期心脏发育过程中,TBX5是心脏传导系统的模式和成熟心肌细胞功能的维持所必需的[11-12]。TBX5编码区的突变在HOS家族聚集患者中的检出率为20%~55%,在散发患者中为15%~40%。TBX5基因中已经发现了超过90个突变,包括错义、无意义、移码突变、剪接突变、复制和染色体重排。HOS的表型既不是由基因型决定,也不是由突变的位置决定。虽然可以出现散发病例,但更常见的是常染色体显性遗传,具有100%的外显率和可变的表达[13],这就解释了虽遗传自母亲,患儿的畸形比母亲更严重。Vanlerberghe等[14]对78例HOS患者进行了最大规模的分子调查,并对表型-基因型进行了相关性分析,发现与错义突变相比,缺失突变者的心脏相关症状较轻。有研究报道无义突变(c.577G>T;p.Gly193*)可引起混合型肺静脉异位引流,也可表现为另一家族的房间隔缺损和(或)室间隔缺损、二叶主动脉瓣和心房颤动[15-16]。尽管在大多数患者中存在家族性传播,但Nourzad等[17]报道30%~40%为新生突变。本文病例为家族聚集性发病,全外显验证为基因的片段缺失覆盖TBX5基因整个Exon9,遗传自母亲,至此患儿及其家族的病症更加明确。
临床上若有相似的表现,还需注意此类患儿需与SALL-4异常如肛门-耳-肢体畸形综合征、Tabatzink手心综合征2型和3型(西班牙语)以及其他类似的上肢疾病,如血小板减少-无桡骨综合征,范可尼贫血、Kaufmann McKusick综合征、Okihiro综合征、Nanger综合征和致畸性胚胎病相鉴别[18-20]。由于其病因、先天性缺陷的表现、预后及遗传方式的不同,鉴别诊断显得很重要, 同时全外显子组测序的应用在其中发挥着决定性作用。不同于大多合并心脏畸形的综合征,HOS患者智力不受影响,生长发育通常是正常的,该患儿及时行手部矫形及先心病手术治疗即可治愈。本例患儿因有家族聚集,临床注意到遗传病可能,但若仅有上肢畸形的患儿,极有可能漏诊,通过查体虽可发现骨骼的异常,但房间隔缺损经常无明显杂音,不做超声心动图很可能被漏诊,有报道患者几十年都未曾诊断该病,就诊时已出现肺动脉高压,气促、呼吸困难、青紫等艾森曼格综合征的不可逆损伤[21],影响患者的生活质量及寿命,有上肢畸形的患者需注意HOS可能。综上,对于上肢畸形合并先心病的患儿,需要注意HOS可能,早期的诊断可以帮助患儿早治疗,随着超声心动技术的提升及孕期检查的普及,通过提高医生对此病的认识,孕期胎儿超声可发现此类患儿,基因检测的完善可以确诊,从而提供生殖抉择,利用产前筛查发现先天性异常,减少畸形患儿的出生。
猜你喜欢
昆明医科大学学报(2022年3期)2022-04-19 13:59:36
宁夏医学杂志(2020年3期)2021-01-21 08:23:42
公民与法治(2020年4期)2020-05-30 12:31:40
天津医科大学学报(2019年6期)2019-08-13 07:04:34
心肺血管病杂志(2019年4期)2019-06-27 07:36:16
文理导航·科普童话(2017年1期)2017-05-31 22:18:57
创新作文(小学版)(2016年11期)2016-11-11 05:45:31
罕少疾病杂志(2016年6期)2016-03-11 16:34:48
西南军医(2016年1期)2016-01-23 02:22:12
实用手外科杂志(2015年2期)2015-08-28 09:50:56
