
黄芪丹参配伍调控ASIC1a/CaMKⅡδ信号抑制酸性微环境中心肌细胞焦亡
2023-03-06 07:46:24张蒙王新东
南京中医药大学学报 2023年2期
张蒙,王新东
(1.南京中医药大学第三临床医学院,江苏 南京 210028;2.上海中医药大学附属曙光医院,上海 201203)
心肌细胞微环境稳态平衡是心脏功能正常的关键因素。冠心病心肌缺血、缺氧状态下发生脂肪酸代谢向糖酵解转变[1],可产生过量乳酸引起细胞外和细胞内pH值显著下降,在急性心肌梗死患者中可降至6.0~6.5[2],导致心肌细胞外酸化,进而形成酸性微环境。细胞外酸化可诱导中性粒细胞活化而促进炎症[3],在炎症相关的心肌损伤中起到重要的作用。因此,阐明缺血缺氧心肌细胞外酸性微环境在心肌炎症中的作用及其潜在的分子机制有助于临床策略的制定和药理学靶点的研究。
酸敏感离子通道(ASICs)是一类细胞外H+活化的阳离子通道,属上皮钠通道(ENaC)/降解蛋白(DEG)超家族[4]。心肌细胞主要表达ASICs亚基ASIC1a。作为细胞膜上的酸受体,ASICs将细胞外微环境的低pH信号传递到细胞中诱导一系列病理变化[5]。ASIC1a可介导细胞外Ca2+流入,Ca2+作为重要的第二信使,参与下游的炎症等病理过程[6-7]。Ca2+/钙调蛋白依赖性蛋白激酶Ⅱ(CaMKⅡ)在缺血损伤的心肌细胞死亡反应中起关键作用[8],CaMKⅡδ是心脏中的主要亚型。细胞焦亡是由caspase-1、caspase-4、caspase-5及其效应子gasdermin家族介导的一种的程序性炎症死亡方式。NLRP3炎症小体的激活是细胞焦亡的经典途径。CaMKⅡδ介导的心肌细胞炎症基因表达和炎症小体激活引发炎症并诱导心肌纤维化重塑[9]。目前尚不清楚ASIC1a是否通过CaMKⅡδ-NLRP3信号参与心肌缺血缺氧后的炎症损伤病理过程。
益气活血是冠心病心肌缺血的经典治法。补气药黄芪和活血药丹参的配伍应用是益气活血法的代表性“药对”,具有抗心肌缺血性重构、调节缺血后能量代谢重编程、抗炎及Ca2+通道调控等效应[10-13]。本研究拟对黄芪丹参配伍调控ASIC1a/CaMKⅡδ信号保护酸性微环境中心肌细胞焦亡的机制进行探讨。
1 材料和方法
1.1 细胞
H9c2大鼠心肌细胞(Procell公司,货号:CL-0089)在专用培养基(Procell公司,货号:CM-0089,主要成分DMEM+10% FBS+1% P/S)中,于37 ℃、5% CO2条件培养。
1.2 主要试剂
胎牛血清(Gibco,货号:C0235),Lactate(迈瑞尔,货号:50-21-5),NS383(MCE公司,货号:HY-131879),Nocistatin Peptide(Abbiotec公司,货号:350301),Annexin V-FITC/PI细胞凋亡检测试剂盒(Sigma公司,货号:APOAF),Fluo 3-AM(Dojindo公司,货号:JU887),逆转录试剂盒、荧光定量检测试剂盒(Takara公司,货号:RR036A、RR820A),PVDF膜(Millipore公司),CCK-8试剂盒、Trizol、BCA蛋白浓度测定试剂盒、ECL化学发光试剂盒(碧云天公司,货号:C0037、R0016、P0012S、P0018FS),ASIC1a抗体(Ptoteintech公司,货号:27235-1-AP),CaMKⅡδ抗体、NLRP3抗体、Caspase-1抗体、GSDMD抗体和GAPDH抗体(Abcam公司,货号:ab300563、ab181052、ab263899、ab138483、ab219800、ab9485)。
1.3 仪器
超净工作台(ESCO公司,型号:ACB-6E1),CO2培养箱(Thermo Fisher Scientific公司,型号:310370),多功能酶标仪(Thermo Fisher Scientific公司,型号:Varioskan LUX),荧光倒置显微镜(Carl Zeiss公司,型号:Multiplex SR-2Y),流式细胞仪(BECKMAN COULTER公司,型号:CytoFLEX),Real-Time PCR循环仪(Bio-Rad公司,型号:CFX Opus Deepwell),电泳仪(Bio-Rad公司,型号:PowerPac),化学发光成像系统(Bio-Rad公司,型号:ChemiDocTMTouch)。
1.4 药物制备
黄芪丹参提取物(HQ)的制备:生黄芪和丹参饮片购自江苏省中西医结合医院中药房。取黄芪6 kg,丹参3 kg,加水热回流提取1.5 h后过滤,重复2次合并滤液,减压浓缩为稠浸膏。用95%乙醇稀释稠浸膏,调节含醇量为70%。静置沉淀后取上清,105 ℃下减压干燥48 h,制成HQ干粉。每1 g HQ干粉相当于5 g处方量。
含HQ的细胞干预药物制备:称取适量的提取物干粉加入PBS于80 ℃恒温水浴锅中加热充分溶解,12 000 r·min-1离心5 min×2次,上清以0.45 μm滤器过滤后再于超净工作台内用0.22 μm滤器将药液过滤除菌,-20 ℃保存备用。
HQ细胞干预药物的浓度测定:分别取按上法制备的HQ细胞干预药物100、500 μL和1 mL置于3支1.5 mL离心管中,放入60 ℃烘箱中48 h充分蒸发水分。将烘干的离心管分别称质量。加药前后2次称取离心管质量的差值即为对应体积的药液的含药量,计算浓度并取平均值。
1.5 CCK-8法检测细胞活力
1.5.1 检测正常培养环境中细胞活力
取对数生长期的细胞,胰酶消化制成细胞悬液,按每孔1×103个接种到96孔板中。细胞分为空白组(Blank,调零组)、对照组(Control)、HQ(0.1、1、10、20、40、60、80、100 μg·mL-1)组。各组均使用H9c2专用培养基模拟正常培养环境,分别加入相应浓度的HQ。培养结束后,使用酶标仪测定吸光度。
1.5.2 检测PH 6.5酸性环境中细胞活力
细胞分为空白组(Blank,调零组)、对照组(Control)、HQ(20、40、60、80μg·mL-1)组。各分组加入相应浓度的HQ处理2 h,除Blank组外,其余组再用 20 mmol·mL-1Lactate处理24 h,维持培养基pH 6.5。培养结束后,每孔加入100 μL CCK-8工作液,使用酶标仪测定450 nm处吸光度。
1.6 细胞分组与干预
选择对数生长期的细胞进行实验。基于细胞活力选择HQ的浓度。将H9c2细胞分为3组:①对照组(Control):细胞正常培养;②pH 6.5酸性环境模型组(Model):细胞用20 mmol·L-1Lactate处理24 h,维持培养基pH 6.5;③HQ+Nocistatin组:60 μg·mL-1HQ预处理细胞2 h,继用1 mmol·L-1Nocistatin预处理1 h,再用20 mmol·L-1Lactate处理24 h;④HQ组:60 μg·mL-1HQ预处理细胞2 h,再用20 mmol·L-1Lactate处理24 h;⑤NS383组:1 μmol·L-1NS383预处理细胞1 h,再用20 mmol·L-1Lactate处理24 h。
1.7 流式细胞术检测细胞凋亡
将细胞分组处理后,用预冷的PBS清洗3次。离心并重悬后,根据试剂盒的说明,用碘化丙啶(PI)和Annexin V-FITC对细胞进行染色。使用流式细胞仪检测细胞凋亡。
1.8 免疫荧光实验
细胞分组干预,PBS冲洗后放入4%多聚甲醛固定20 min,随后加入0.5% Triton X-100通透15 min,滴加5%BSA均匀覆盖细胞,室温条件下进行封闭30 min。滴加一抗,将细胞放入湿盒中,恒温4 ℃的条件下孵育过夜。室温环境中荧光二抗(Cy3标记)孵育50 min。DAPI染液室温孵育10 min后用荧光倒置显微镜进行图像采集。
1.9 qPCR实验
用TRIzol试剂从各组细胞中提取总RNA,测定RNA浓度。按逆转录试剂盒说明书将RNA逆转录为cDNA。使用PCR系统进行实时反应。以GAPDH为内参,计算各组相应基因的2-ΔΔCt值。引物序列表1所示。

表1 特定引物序列Table 1 Specific primer sequences
1.10 Western blot分析
使用裂解缓冲液从不同组的H9c2细胞中提取总蛋白并通过BCA定量。将蛋白质加载到SDS-PAGE中并通过电泳分离。将分离的蛋白质转移到PVDF膜上。在TBST中用5%脱脂牛奶封闭1 h后,将膜与一抗(ASIC1a,1∶1 000;CaMKⅡδ,1∶1 000;NLRP3,1∶1 000;Caspase-1,1∶1 000;GSDMD,1∶1 000)在4 ℃下孵育过夜。用TBST洗涤膜,并与相应的二抗(1∶5 000)在室温下孵育。运用化学发光法检测蛋白质条带,Image J软件分析。
1.11 统计学方法
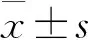
2 结果
2.1 HQ促进pH 6.5酸性环境中降低的细胞活力
细胞在正常的培养环境中与Control组相比,HQ在0.1~100 μg·mL-1对细胞活力无明显影响(图1A)。在pH 6.5酸性环境中与Blank组相比,细胞活力显著降低(P<0.01);与Control组比较,HQ对细胞活力具有显著促进作用,尤其是60、80 μg·mL-1浓度组(P<0.01),且60 μg·mL-1优于80 μg·mL-1浓度组(P<0.05),因此本研究选用60 μg·mL-1浓度作为细胞干预剂量(图1B)。此结果提示HQ具有保护pH 6.5酸性环境中细胞凋亡的作用。
注:与Blank组相比,**P<0.01;与Control组相比,##P<0.01;与HQ 60 μg·mL-1组相比,▲图1 CCK8检测细胞活力Fig.1 CCK8 assay for cell viability
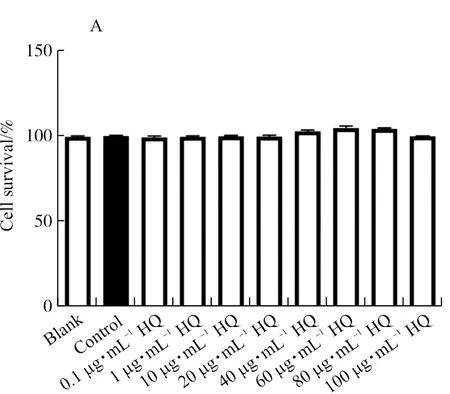
2.2 HQ抑制pH 6.5酸性环境中心肌细胞凋亡
本研究进一步采用流式细胞术检测pH 6.5酸性环境中心肌细胞凋亡,结果显示:pH 6.5酸性环境可诱导细胞凋亡明显增加(P<0.01),HQ和ASIC1a抑制剂NS383均可减少酸性环境中的心肌细胞凋亡(P<0.01),且HQ的这种保护效应可被ASIC1a激动剂Nocistatin逆转,提示HQ对酸性环境中的心肌细胞凋亡的保护效应与调控ASIC1a酸敏感Ca2+通道有关(图2)。
注:Q1-UL.坏死细胞;Q1-UR.晚期凋亡细胞;Q1-LL.正常细胞;Q1-LR.早期凋亡细胞。与Control组相比,**P<0.01;与Model组相比,##P<0.01;与HQ组相比,图2 流式细胞术检测各组pH 6.5酸性环境中心肌细胞凋亡Fig.2 Flow cytometry detection of cardiomyocyte apoptosis in the acidic environment of pH 6.5 in each group
2.3 HQ抑制pH 6.5酸性环境中心肌细胞ASIC1a表达
为明确HQ对心肌细胞酸敏感离子通道ASIC1a的影响,本研究采用免疫荧光检测pH 6.5酸性环境中心肌细胞中ASIC1a表达情况,结果显示:pH 6.5酸性环境可诱导ASIC1a蛋白表达明显增加(P<0.01),HQ和ASIC1a抑制剂NS383均可明显降低酸性环境中ASIC1a表达(P<0.01),且HQ的这种保护效应可被ASIC1a激动剂Nocistatin逆转,提示HQ对酸性环境中心肌细胞的抑制效应与调控ASIC1a有关(图3)。
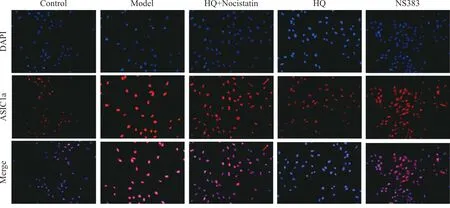
注:比例尺=50 μm。与Control组相比,**P<0.01;与Model组相比,##P<0.01;与HQ组相比,▲
2.4 HQ下调pH 6.5酸性环境中心肌细胞NLRP3、Caspase-1、IL-1β及GSDMD mRNA表达
本研究分析了NLRP3炎症小体介导的细胞焦亡信号相关基因mRNA表达,结果显示:pH 6.5酸性环境可诱导H9c2心肌细胞NLRP3、Caspase-1、IL-1β及GSDMD mRNA表达明显增加(P<0.01),HQ和ASIC1a抑制剂NS383均可显著下调酸性环境中的以上基因mRNA表达(P<0.01),且HQ的下调效应可被ASIC1a激动剂Nocistatin逆转,提示HQ对酸性环境中的心肌细胞损伤的保护效应与抑制NLRP3炎症小体介导的细胞焦亡有关(图4)。
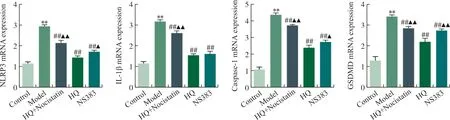
注:与Control组相比,**P<0.01;与Model组相比,##P<0.01;与HQ组相比,▲图4 pH 6.5酸性环境中心肌细胞NLRP3、Caspase-1、IL-1β及GSDMD mRNA表达比较Fig.4 Comparison of cardiomyocytes mRNA expression of NLRP3, Caspase-1, IL-1β and GSDMD in the acidic environment of pH 6.5
2.5 HQ下调pH 6.5酸性环境中心肌细胞ASIC1a、CaMKⅡδ、NLRP3、Caspase-1、Cleaved Caspase-1、GSDMD及GSDME-N蛋白表达
Western blot分析各组心肌细胞ASIC1a、CaMKⅡδ、NLRP3、Caspase-1和GSDMD蛋白表达的结果显示:pH 6.5酸性环境可诱导心肌细胞ASIC1a、CaMKⅡδ、NLRP3、Caspase-1、Cleaved Caspase-1、GSDMD及GSDME-N蛋白表达明显增加(P<0.01),HQ和ASIC1a抑制剂NS383均可显著下调酸性环境中的以上基因蛋白表达(P<0.05,P<0.01),且HQ对以上蛋白的下调效应均可被ASIC1a激动剂Nocistatin逆转(P<0.05,P<0.01),提示HQ对酸性环境中的心肌细胞损伤的保护效应与调控ASIC1a介导的CaMKⅡδ表达和相关的抑制NLRP3炎症小体介导的细胞焦亡有关(图5)。
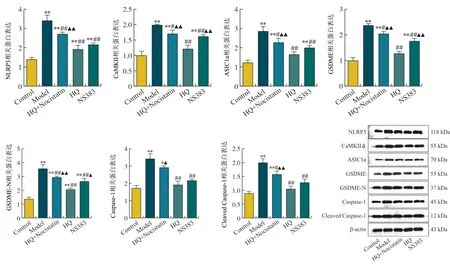
注:与Control组相比,**P<0.01;与Model组相比,#P<0.05,##P<0.01;与HQ组相比,▲图5 pH 6.5酸性环境中心肌细胞ASIC1a、CaMKⅡδ、NLRP3、Caspase-1、Cleaved Caspase-1、GSDMD及GSDME-N蛋白表达比较Fig.5 Comparison of cardiomyocytes protein expression of ASIC1a, CaMKⅡδ, NLRP3, Caspase-1, Cleaved Caspase-1, GSDMD and GSDME-N in the acidic environment of pH 6.5
3 讨论
乳酸和相关的H+离子可在冠心病心肌组织缺氧或在能量代谢从氧化磷酸化重编程为糖酵解的过程中产生。在缺氧的情况下,大部分丙酮酸转化为乳酸,由乳酸转运蛋白单羧酸转运蛋白4(MCT4)向外转运到细胞外环境,进而形成酸性微环境[14]。酸性微环境是缺血缺氧心肌在能量代谢重编程的状态下形成的细胞微环境,对心肌细胞的存活和损伤可产生重要影响,但具体机制仍有待阐释。心肌细胞炎性损伤是缺血缺氧的关键结果,乳酸作为糖酵解的产物在诱导NLRP3炎症小体的激活导致炎症损伤方面发挥重要作用[15]。黄芪丹参配伍作为冠心病的经典治疗药对,临床疗效确切,我们前期的研究结果提示其部分机制与调节缺血缺氧心肌的能量代谢重编程有关[13]。在本研究中我们进一步证实了黄芪丹参提取物可调节酸性微环境下心肌细胞的细胞焦亡相关的炎症损伤,并且其机制与调控酸敏感离子通道ASIC1a介导的CaMKⅡδ信号有关。
乳酸产生、酸中毒和缺氧通常被认为是心肌血流减少或阻断后的重要病理进程。同时,高乳酸浓度还与缺血或衰竭心肌的能量重构(代谢模式从氧化磷酸化转变为糖酵解),如糖酵解、增加的谷氨酰胺分解和血管的低灌注率等也有密切关联,导致乳酸在非缺氧区域的积累。乳酸不仅是心肌细胞所处微环境酸化的主要参与者,而且可作为“信号分子”的重要作用,参与心肌细胞存活和损伤的不同机制。因此,针对乳酸、酸性微环境及其相关的能量代谢重编程的调节是保护缺血缺氧心肌的重要临床策略。NLRP3炎性小体由NOD样受体蛋白3(NLRP3)、凋亡相关斑点样衔接蛋白(ASC)和Caspase-1效应蛋白组成,ASC连接上游的NLRP3和下游的Caspase-1。激活的NLRP3炎性小体参与由细胞焦亡引起的细胞损伤,GSDMD是细胞焦亡核心执行蛋白。从本研究的结果可以看出,在乳酸诱导的pH 6.5酸性环境中心肌细胞活力显著降低、凋亡明显增加,NLRP3、Caspase-1、IL-1β、GSDMD等NLRP3炎症体介导的细胞焦亡信号也明显增加,提示酸性微环境对心肌细胞产生炎性损伤影响。而给予HQ干预可明显保护心肌细胞减轻酸性微环境诱导的细胞凋亡和焦亡损伤,显示了HQ对酸性微环境下心肌的保护效应。
针对酸性微环境是从何种途径诱导细胞焦亡炎性损伤的机制,我们进一步从酸敏感离子通道ASIC1a介导的CaMKⅡδ信号的视角开展了研究。结果显示,在pH 6.5酸性微环境下ASIC1a明显增加,提示ASIC1a介导的胞内Ca2+作为第二信使参与了下游的焦亡损伤信号。心肌细胞Ca2+超载是缺血损伤的重要因素,可通过CaMKⅡ等下游信号引发一系列心肌细胞死亡和心肌功能障碍事件。CaMKⅡδ是心脏中的主要亚型,是药物抑制的潜在靶点。持续的CaMKⅡ激活诱发程序性细胞死亡,包括细胞凋亡和坏死性凋亡,是其有害影响的关键潜在机制之一。CaMKⅡ整合β-肾上腺素能、Gq偶联受体、活性氧(ROS)、高血糖、NLRP3炎症小体[16]和促死亡细胞因子信号传导[17],并加重不良代谢重编程[18]。药理学抑制CaMKⅡδ可保护缺血心肌线粒体凋亡和炎症[19]。本研究结果显示,在pH6.5酸性微环境下,伴随ASIC1a表达增加,CaMKⅡδ的蛋白表达也明显增加,提示CaMKⅡδ可能作为中转信号介导了酸性微环境-ASIC1a与细胞焦亡间的串话。HQ可抑制酸性微环境下ASIC1a和CaMKⅡδ激活,并且上述效应可被ASIC1a激动剂逆转,提示HQ对酸性微环境中心肌细胞焦亡的抑制效应与调控ASIC1a/CaMKⅡδ信号有关。
综上所述,本研究发现pH6.5酸性微环境下心肌细胞发生凋亡和NLRP3炎症小体激活介导的细胞焦亡与酸敏感离子通道ASIC1a介导的CaMKⅡδ信号激活有关,HQ可通过下调酸性微环境中ASIC1a/CaMKⅡδ信号保护心肌细胞免受凋亡和细胞焦亡损伤。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:24:52
医学综述(2022年7期)2022-04-19 12:31:12
云南化工(2021年10期)2021-12-21 07:33:28
服饰导报·鞋世界(2021年4期)2021-05-17 14:01:41
昆明医科大学学报(2020年12期)2021-01-26 00:44:42
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:26
福建基础教育研究(2019年8期)2019-05-28 08:39:51
中国药理学通报(2019年5期)2019-01-11 18:03:39
中学生数理化·八年级物理人教版(2017年6期)2017-11-09 06:00:43
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
