
基于生物信息学挖掘骨关节炎潜在的关键生物标志物*
2023-03-01 12:00戈昱凡缪逸鸣
国际检验医学杂志 2023年4期
应 璞,路 通,许 岳,周 烨,戈昱凡,薛 燚,缪逸鸣
南京中医药大学常熟附属医院骨科,江苏常熟 215500
骨关节炎(OA)是一种临床上最常见的以关节软骨退变和破坏为特征的慢性关节疾病,其临床表现为关节疼痛、僵硬、肿胀伴关节活动受限等[1-2]。OA发病机制复杂,涉及关节软骨的机械刺激及退变、滑膜组织的病变、软骨下骨微循环改变、炎症反应等[3-5],但目前OA的具体机制仍未阐明。近年来,随着微阵列技术和生物信息学的发展,通过对OA的高通量微阵列和基因芯片数据进行研究,越来越多的OA相关差异表达基因(DEGs)被发现[6-9]。这些基因的发现,有望为阐明OA发病的分子机制和治疗靶点提供新的理论指导。本研究从基因表达综合数据库(GEO)下载了mRNA数据集和微小RNA(miRNA)数据集,选取OA滑膜组和正常滑膜组进行比较与分析,分析OA中差异表达的mRNA和miRNA。本研究旨在通过生物信息学方法筛选出与OA发生、发展相关的关键mRNA和miRNA,建立mRNA-miRNA共表达网络,从而进一步探索OA的发病机制和潜在生物标志物。
1 材料与方法
1.1材料 本研究采用GE数据库(https://www.ncbi.nlm.nih.gov/geo/)下载mRNA数据集GSE55457、GSE82107、GSE55235和miRNA数据集GSE143514。其中GSE55457数据集包括10例OA滑膜组织和10例正常滑膜组织,GSE82107数据集包含10例OA滑膜组织和7 例正常滑膜组织,GSE55235数据集包含26例OA滑膜组织和20例正常滑膜组织,GSE143514数据集包含5例OA滑膜组织和3例正常滑膜组织。
1.2方法
1.2.1差异基因筛选 所有芯片数据由R语言软件的affy程序包预处理,随后通过limma程序进行筛选,OA与正常滑膜之间的差异表达基因(DEGs),按log2FC>1且P<0.05的标准进行筛选(FC为差异倍数)。重叠差异表达基因数据集采用R语言软件的VennDiagram包Venn图显示。
1.2.2差异基因GO和KEGG富集分析 利用在线分析工具Metascape(http://metascape.org/)对差异表达基因进行GO功能富集分析和KEGG通路富集分析,以P<0.05且基因计数≥5为差异有统计学意义。
1.2.3蛋白质-蛋白质相互作用(PPI)网络分析及miRNA-mRNA共表达网络分析 利用在线分析数据库STRING ( http://string-db.org/)将筛选出的差异表达基因导入数据库,进行差异基因PPI网络分析,并利用Cytoscape软件(https://cytoscape.org)构建PPI网络。
2 结 果
2.1筛选差异表达的mRNA和miRNA 对mRNA数据集GSE55457、GSE82107和miRNA数据集GSE143514原始数据进行处理后,按筛选标准进行分析,最后从GSE55457中筛选出626个差异表达mRNA,其中313个上调、313个下调。按同样的筛选标准从GSE82107中筛选出1 647个差异表达mRNA,其中498个下调、1 147个上调,从GSE143514中筛选出142个差异表达miRNA,其中22个下调、120个上调,见图1。随后,利用Venn图比较GSE55457和GSE82107两个数据集的差异表达mRNA,得到99个相同的mRNA,见图2。

注:A为差异表达mRNA数据集GSE55457;B为差异表达mRNA数据集GSE82107;C为差异表达miRNA数据集GSE143514。
2.2鉴定相关的潜在mRNA 首先对数据集GSE55235的数据进行标准化处理,然后通过limma软件包从GSE55235中筛选出334个差异表达的mRNA,见图3。为了使网络符合无尺度网络分布,利用权重基因共表达网络分析(WGCNA)软件包中的函数pickSoftThreshold计算权重值,并选择软阈值β=16用来构建共表达网络,见图4A,并采用平均连锁层次聚类方法对基因进行聚类,见图4B。同时,对模块进行聚类分析,然后根据各模块特征基因与样本类型的相关性,发现绿色模块与样本类型的相关性最大,见图4C。这些结果提示绿色模块中的27个基因可能与OA的发生密切相关。此外,比较绿色模块中筛选得到的27个mRNA与图2中筛选得到的99个mRNA,最终获得OA的19个交叉的关键mRNAs和27个绿色模块mRNAs,见图4D。19个交叉的关键mRNA为APOLD1、CD52、趋化因子配体(CXCL)13、CXCL2、CXCL3、DUSP1、FKBP5、FOSB、IGJ、IGLL5、JUN、基质金属蛋白酶(MMP)1、ZB1、NFIL3、NKG7、NR4A2、磷脂酶Cδ3(PLCD3)、POU2AF1、TNFRSF17。和27个绿色模块mRNAs为BCL6、RHOB、NFIL3、KLF4、PLCD3、CEBPD、GADD45A、BTG1、GADD45B、EIF1、APOLD1、CD52、NKG7、CXCL13、CXCL3、CXCL2、DUSP1、MMP1、JUN、FKBP5、NR4A2、FOSB、IGJ、TNFRSF17、POU2AF1、MZB1、IGLL5。
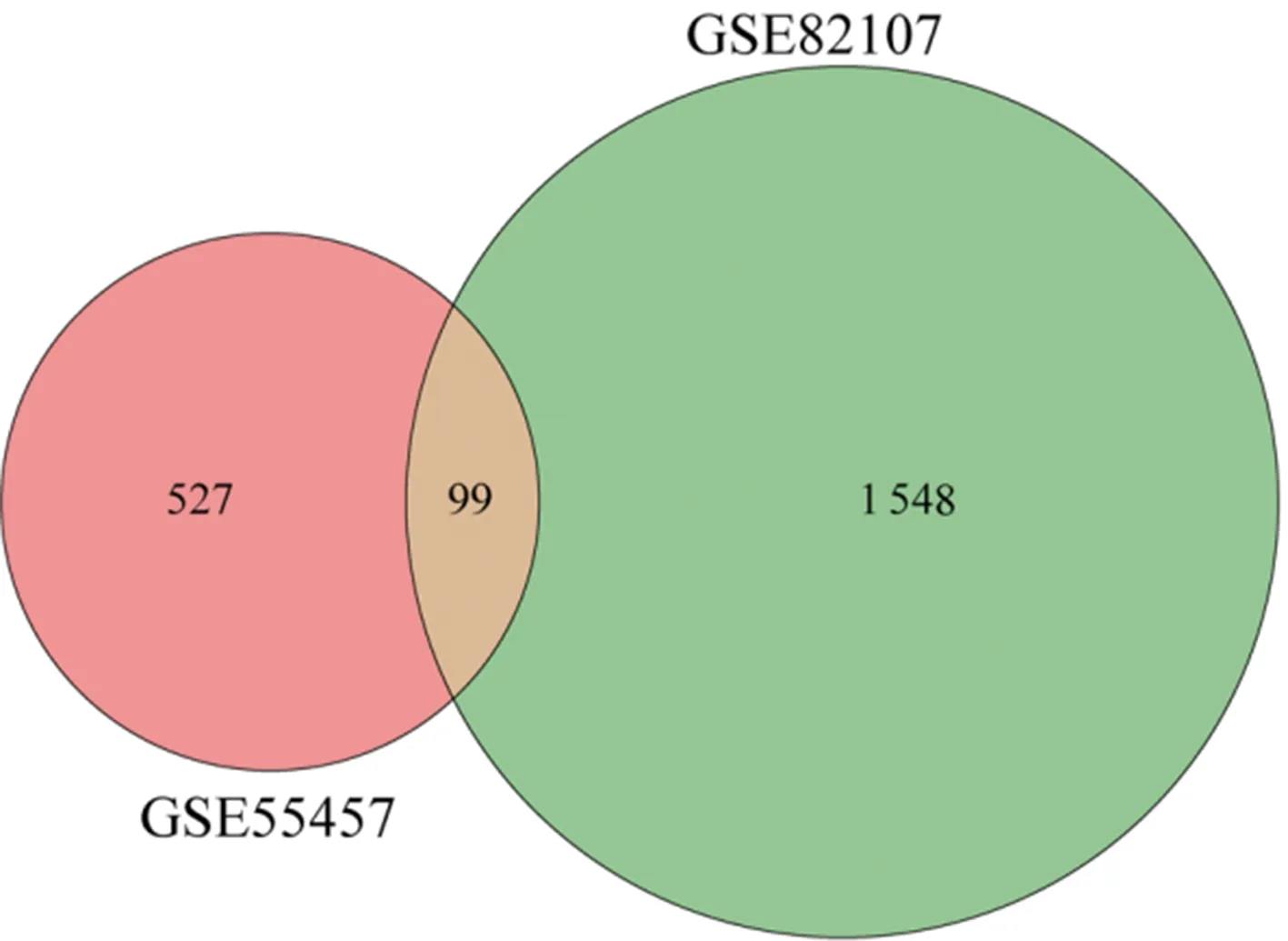
图2 GSE55457和GSE82107共同差异表达mRNA的Venn图
2.3mRNA的GO分析和KEGG分析 GO功能富集分析显示,19个关键mRNAs主要参与免疫相关过程。例如体液免疫反应、细胞对趋化因子的反应、白细胞趋化、粒细胞趋化、粒细胞迁移、细胞对脂多糖的反应、B细胞活化和白细胞迁移、抗菌体液反应以及对细菌的防御反应等,见图5A。KEGG通路富集分析显示,19个关键mRNAs主要参与白细胞介素(IL)-17信号通路、肿瘤坏死因子(TNF)信号通路、转化生长因子-β(TGF-β)信号通路、NOD样受体信号通路、趋化因子信号通路、白细胞介素信号通路和PI3K/AKT信号通路等,见图5B。
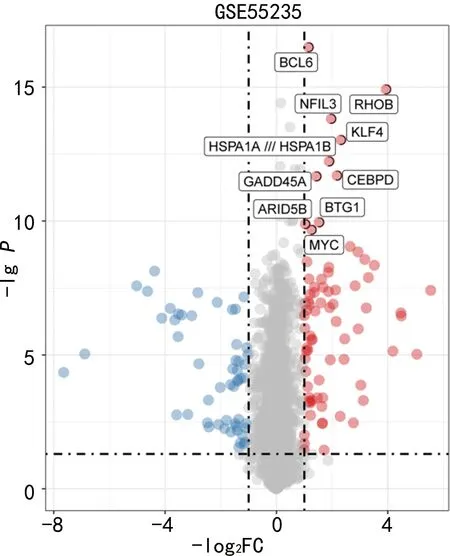
图3 OA中差异表达mRNA数据集GSE55235火山图
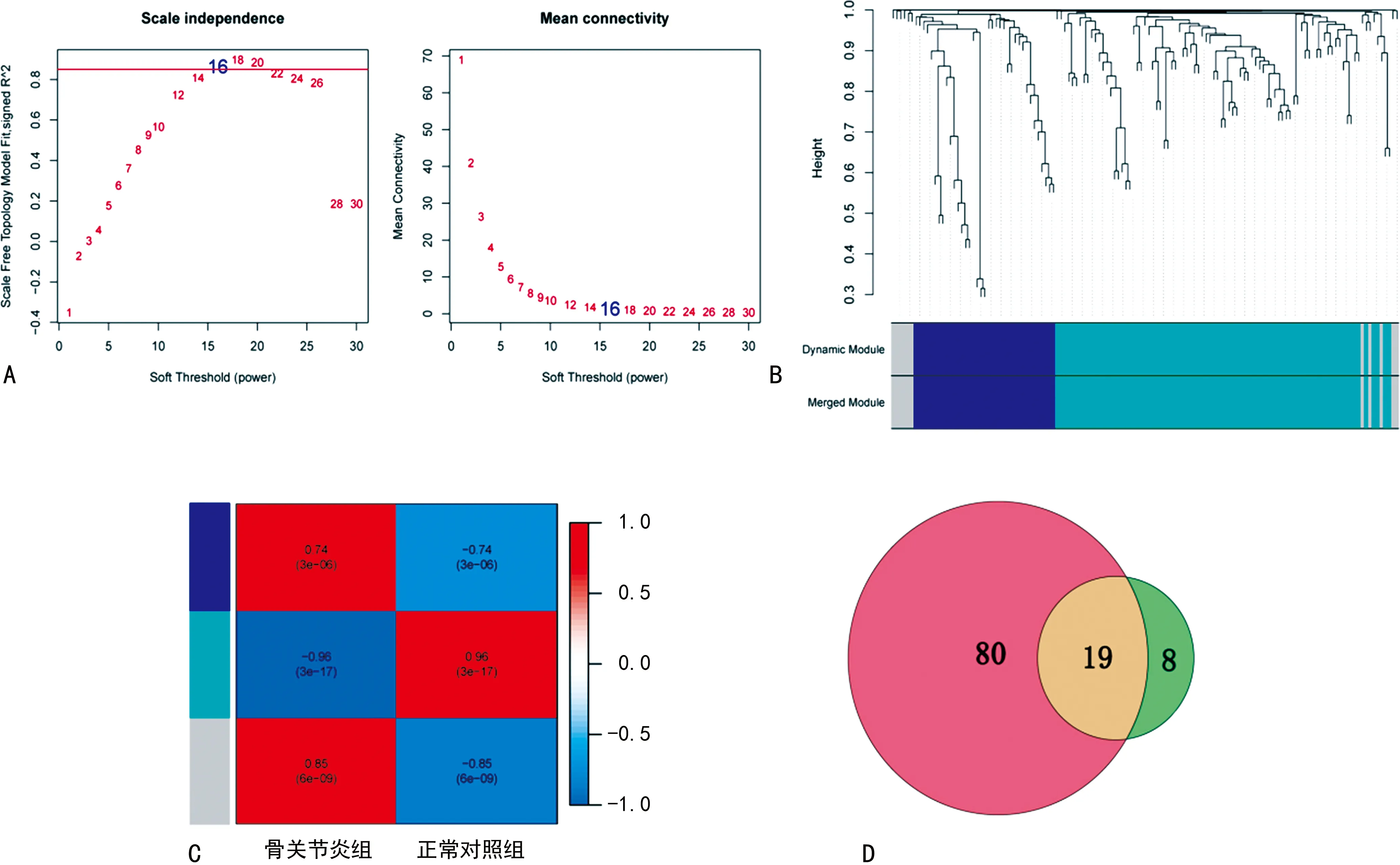
注:A为各种软阈值功率的网络拓扑分析;B为基因模块的聚类树状图;C为OA和正常样本之间基因模块的相关性分析;D为图2中99个筛选得到的mRNA和27个绿色模块筛选得到的共同差异表达mRNA的Venn图。
2.4PPI网络分析及miRNA-mRNA共表达网络分析 利用STRING数据库和Cytoscape软件构建一个由11个节点和24条边组成的PPI网络,见图6A。随后通过MCODE插件获得PPI评分最高的子网络,该子网络由CXCL2、JUN、CXCL3、MMP1、PLCD3共5个mRNA组成,见图6B,说明CXCL2、JUN、CXCL3、MMP1、PLCD3这5个基因可能是OA最关键的基因。同时利用DIANA Tools在线数据库(http://diana.imis.athena-innovation.gr/DianaTools/index.php)分析预测5个关键基因的靶向miRNA。然后,将获得的所有靶向miRNA与从GSE143514中筛选出142个差异表达miRNA,见图1C,进行比较,获得5个关键基因的24个共同miRNA,见图6C。此外,还预测了39个miRNA-mRNA基因对。根据预测结果,利用Cytoscape软件构建了一个由29个节点和39条边组成的mRNA-miRNA共表达网络,见图6D、表1。

注:A为GO功能富集分析;B为KEGG功能富集分析。

表1 OA的mRNA-miRNA共表达网络中的miRNAs和mRNAs
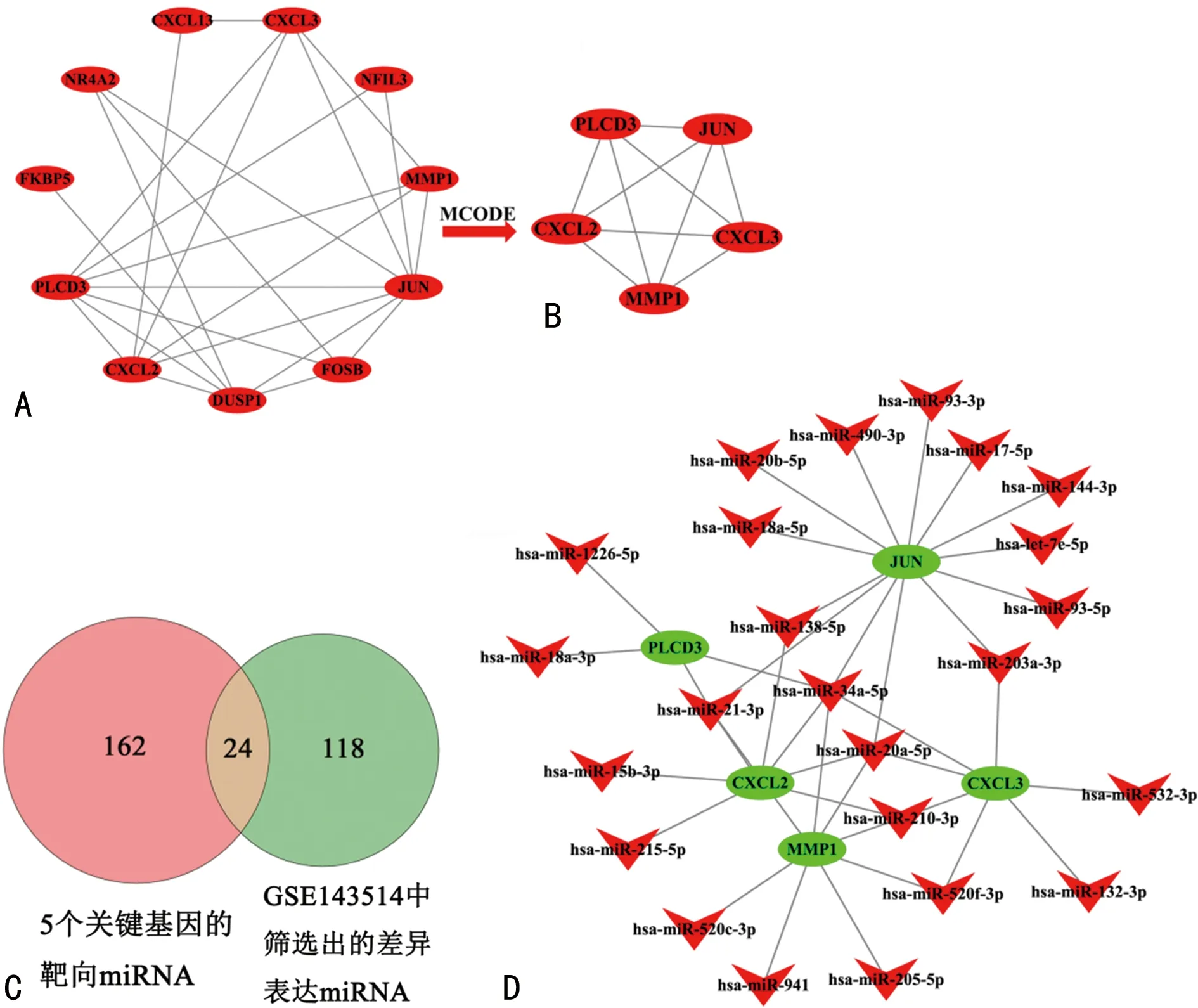
注:A为PPI网络;B为PPI网络5个mRNA组成;C为靶向miRNA的差异表达筛选;D为mRNA-miRNA共表达网络。
3 讨 论
OA发病机制复杂,目前已有多个基因和miRNA发现参与OA的发生、发展,但其发病机制仍未被阐明[10-14]。基于高通量测序基因芯片技术和生物信息学技术的发展,能够筛选出OA的潜在的关键基因和关键miRNA,从而深入研究其分子机制并提供潜在的治疗靶点。
本研究整合了4个GEO数据集,并通过筛选OA和健康对照组的滑膜组织中差异表达的mRNA和miRNA,确定了19个关键mRNA。通过GO功能富集分析和KEGG通路分析,这些关键mRNA主要参与免疫相关过程,例如体液免疫反应、细胞对趋化因子的反应、白细胞趋化、粒细胞趋化等,主要参与IL-17信号通路、TNF信号通路、TGF-β信号通路等。HAN等[15]通过研究5个GEO数据库获得了5个OA的关键差异表达基因(TLR7、CSF1R、APOE、C1QA、CCL5),这些基因广泛参与炎症反应调控、血管形态发生、内皮细胞迁移、体液免疫应答等生物学过程。MEEHAN等[16]证实OA患者中关节滑液中的IL-4、IL-6、IL-8、IL-15水平显著高于血清中的水平,证实OA患者中的滑液中存在大量趋化因子和促炎细胞因子。目前的研究认为,与OA相关的促炎因子,包括IL-1β、TNF-α、IL-6、IL-15、IL-17和IL-18,以及减少OA相关的关键抗炎因子,包括IL-4、胰岛素样生长因子和TGF-β等[3]。这些研究的结果与本研究结果是一致的,证实了炎症和相关免疫反应在OA的发生、发展中有着重要的作用。
本研究通过构建PPI网络分析获得的5个关键基因分别为CXCL2、CXCL3、JUN、MMP1、PLCD3。其中CXCL2和CXCL3都是趋化因子CXC亚家族成员,均为炎性趋化因子,对多形核白细胞具有趋化作用。STONE等[17]发现,OA半月板细胞中趋化因子(CXCL1、CXCL2)的表达显著增加,半月板细胞的促炎刺激增加了基质金属蛋白酶的产生和分解代谢基因的表达,在关节损伤后OA的发展中发挥重要作用。LYNCH等[18]评估了43种生物标志物,结果发现OA软骨组织中趋化因子CXCL3显著高于正常软骨组织。于雅丽等[19]发现JUN基因被确定为有价值的OA生物标记物,这与本研究结果一致。c-JUN蛋白是一种转录因子,各种物理、化学、生物学刺激及细胞应激反应都可以通过促进c-JUN蛋白表达活化,进而对细胞增殖、凋亡等生物学过程进行调控。XU等[20]发现膝OA患者的软骨下骨细胞中ERK/c-JUN N-末端激酶(JNK)信号通路被激活,并参与调控成骨,促进膝OA的发生、发展。目前已有多项研究表明软骨组织或滑液中MMP-1的高水平可能与膝关节OA的发病密切相关[16,21-22]。PLCD3其被发现能够促进肿瘤细胞的增殖、迁移和侵袭[23],然而在OA的发病机制中的作用较少报道,需要后续更多研究探讨PLCD3在OA致病机制中的作用。
本研究共发现39对mRNA-miRNA共表达网络,其中miR-34a-5p在5组mRNA-miRNA均出现,提示其可能在OA的发病机制中存在重要作用。SONG等[24]指出,下调miR-34a-5p可促进OA细胞增殖,抑制细胞凋亡和自噬;ENDISHA等[25]也指出,miR-34a-5p可以促进OA期间的关节破坏。这些研究表明,miR-34a-5p是OA细胞表达的潜在调控因子[24-26]。另外,miR-20a-5p在4组mRNA-miRNA均出现,提示其可能在OA的致病机制中也存在重要作用[27]。ROSS等[28]通过整合mRNA和miRNA测序数据,创建了miRNA-mRNA相互作用网络,发现miR-20a-5p可以显著降低降解酶的产生以及炎症相关基因的表达。这些miRNA的发现,为研究OA的分子机制提供了方向,也为OA的治疗提供了潜在的靶点。
综上所述,本研究通过生物信息学的方法发现了OA与炎症及相关免疫反应存在密切联系。同时,也发现了5个可能参与OA发病机制的基因:CXCL2、CXCL3、JUN、MMP1、PLCD3。另外,还发现了一些OA相关的mRNA-miRNA共表达网络,并提出miR-34a-5p、miR-20a-5p可能会成为未来研究OA分子机制和治疗OA的潜在靶点。
猜你喜欢
舰船科学技术(2022年10期)2022-06-17
安徽农业科学(2022年1期)2022-02-14
建材发展导向(2021年14期)2021-08-23
昆明医科大学学报(2020年12期)2021-01-26
中国组织化学与细胞化学杂志(2016年3期)2016-02-27
中国学术期刊文摘(2016年1期)2016-02-13
中国学术期刊文摘(2016年1期)2016-02-13
中国医药生物技术(2015年4期)2015-12-26
中华胰腺病杂志(2015年5期)2015-12-08
现代检验医学杂志(2015年4期)2015-02-06
