
Inhibition of autophagy rescues HT22 hippocampal neurons from erastin-induced ferroptosis
2023-02-24 05:25NoraHankeAbdelhaqRami
中国神经再生研究(英文版) 2023年7期
Nora Hanke,Abdelhaq Rami
Abstract Ferroptosis is a regulated form of cell death which is considered an oxidative iron-dependent process.The lipid hydroperoxidase glutathione peroxidase 4 prevents the iron (Fe2+)-dependent formation of toxic lipid reactive oxygen species.While emerging evidence indicates that inhibition of glutathione peroxidase 4 as a hallmark of ferroptosis in many cancer cell lines,the involvement of this biochemical pathway in neuronal death remains largely unclear.Here,we investigate,first whether the ferroptosis key players are involved in the neuronal cell death induced by erastin.The second objective was to examine whether there is a cross talk between ferroptosis and autophagy.The third main was to address neuron response to erastin,with a special focus on ferritin and nuclear receptor coactivator 4-mediated ferritinophagy.To test this in neurons,erastin (0.5–8 µM) was applied to hippocampal HT22 neurons for 16 hours.In addition,cells were cultured with the autophagy inhibitor,3-methyladenin (10 mM) and/or ferroptosis inhibitors,ferrostatin 1 (10–20 µM) or deferoxamine (10–200µM) before exposure to erastin.In this study,we demonstrated by immunofluorescence and western blot analysis,that erastin downregulates dramatically the expression of glutathione peroxidase 4,the sodium-independent cystine-glutamate antiporter and nuclear receptor coactivator 4.The protein levels of ferritin and mitochondrial ferritin in HT22 hippocampal neurons did not remarkably change following erastin treatment.In addition,we demonstrated that not only the ferroptosis inhibitor,ferrostatin1/deferoxamine abrogated the ferroptotic cell death induced by erastin in hippocampal HT22 neurons,but also the potent autophagy inhibitor,3-methyladenin.We conclude that (1) erastin-induced ferroptosis in hippocampal HT22 neurons,despite reduced nuclear receptor coactivator 4 levels,(2) that either nuclear receptor coactivator 4-mediated ferritinophagy does not occur or is of secondary importance in this model,(3) that ferroptosis seems to share some features of the autophagic cell death process.
Key Words:erastin;ferritin;ferritinophagy;ferroptosis;glutathione peroxidase 4;HT22 neurons;nuclear receptor coactivator 4
Introduction
Ferroptosis is an iron-dependent and oxidative damage-related form of regulated cell death,which is morphologically,biochemically,and genetically different from other forms of cell death (Dixon et al.,2012).It has been reported that cellular iron overload leads to increased reactive oxygen species(ROS) and accumulation of lipid peroxidation in many cell types (Dixon et al.,2014;Gao and Dixon 2016;Stockwell et al.,2017).In addition,ferroptosis can be prevented by glutathione peroxidase 4 (GPX4) and ferroptosis inhibitors,such as ferrostatin 1 (Fer-1) and the iron chelator deferoxamine (Dixon et al.,2012;Angeli et al.,2014,2017;Gao and Dixon,2016;Yang and Stockwell,2016;Li et al.,2017).
Ferroptosis can be induced chemically by erastin,which inhibits the glutamate/cystine antiporter,and subsequently suppresses cellular cystine uptake and depletes glutathione.Glutathione is fundamental in maintaining the redox balance and defending against oxidative stress,including reactive oxygen species (Kim et al.,2015).Erastin has been widely found to induce ferroptosis in several types of cancer cells (Jiang et al.,2015;Zhao et al.,2020)However,the involvement of these erastin-related biochemical pathways in neuronal death remains largely unclear.Under oxidative stress conditions,cells must actively regulate their iron-carrier proteins such as transferrin,lactotransferrin (Gao et al.,2016).Transferrin is an iron-carrier protein that can be transported into the cell via receptor-mediated endocytosis (Andrews and Schmidt 2007).Lactotransferrin is known as an iron-binding protein.Mechanisms controlling iron-loading of transferrin and or lactotransferrin may impact ferroptosis.On the other hand,cells must buffer their intracellular iron by using cytosolic iron storage proteins such as NCOA4 and ferritins.Mitochondrial ferritin (mtFt) and H-chain ferritin (FTH) are the most known iron storage proteins in the brain.Accumulating evidence indicates the involvement of mtFt in some neurodegenerative diseases,especially Alzheimer´s disease and Parkinson´s disease (Guiney et al.,2017;Tang et al.,2018).Mitochondrial ferritin seems to have inhibitory effects on oxidative stress-dependent neuronal damage and acts as a neuroprotective factor to maintain normal neuronal function (Shi et al.,2010;Guan et al.,2017).Previous studies suggested that overexpression of mtFt may prevent cytosolic iron accumulation and protect neuroblastoma cells from oxidative stress(Gao and Chang,2014;Hassannia et al.,2018).In addition,it has been shown that ferritin is a source of iron and protection from iron-induced toxicities(Reif,1992;Aust,1995).Mouse brain deficient in ferritin exhibited increased evidence of oxidative stress (Thompson et al.,2003) and oligodendrocytes provide anti oxidant functions for neurons by secreting ferritin (Mukherjee et al.,2020).The ferritin turnover is checked by an autophagic cargo receptor,NCOA4,which facilitates the delivery of ferritin for lysosomal degradation via a selective autophagy process,called ferritinophagy (Mancias et al.,2014;Quiles and Mancias 2019).It has been reported that ferriti nophagy is involved in ferroptosis and that the genetic deletion of NCOA4 inhibits ferriti nophagy and blocks lipid peroxidation and ferroptosis by decreasing the levels of bioavailable intracellular iron (Gao et al.,2016).
Based on the link between NCOA4 and ferritin in regulating intracellular iron levels,we intend to investigate,first whether the ferroptosis key players are involved in the neuronal cell death induced by erastin.The second objective was to examine whether there is a cross talk between ferroptosis and autophagy.The third main was to address neuron response to erastin,with a special focus on ferritin and NCOA4-mediated ferriti nophagy.
Methods
Mouse hippocampal neurons (HT22) cells were kindly provided by The Salk Institute for Biological Studies,La Jolla,CA,USA.HT-22 is an immortalized mouse hippocampal cell line subcloned from the HT-4 cell line.The parental HT-4 cell line was derived from the immortalization of mouse neuronal tissues with a temperature-sensitive SV40 T-antigen.HT22 neuronal cell line is a suitable model for studying oxidative stress due to deficiency of N-methyl-Daspartate receptor (Sagara and Schubert 1998).
Drug treatment
The ferroptosis inducer erastin (Cat# HY-15763,Hvcultec,Beutelsbach,Germany),Fer-1 (ferroptosis inhibitor,Cat# ab146169,Abcam,Berlin,Germany),and 3-methyladenin (3-MA [autophagy inhibitor],Sigma,Taufkirchen,Germany) were dissolved in dimethyl sulfoxide (DMSO;Sigma).Deferoxamine (DFO,ferroptosis inhibitor,Sigma) was dissolved in deionized water.Cells were seeded in 96-well plates for viability assay,24-well plates for immunofluorescence staining,and western blot assay.To assess the opti mal concentration of erastin,hippocampal HT22 neurons were exposed to the drug at 0.3,0.4,0.5,0.6,0.7,and 0.8 µM for 12 hours.
Different concentrations of Fer-1 (10 and 20 µM) and DFO (100 and 200µM) were applied to hippocampal HT22 neurons after 12 hours of erastin treatment (0 and 5 µM).The opti mal experimental conditions occurred after incubation with 20 µM for Fer-1 and 100 µM for DFO.After 6,12,and 24 hours,the subsequent analyses were conducted.
Cell viability detection
The detection of cell viability was performed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT,Merck,Taufkirchen,Deutschland)assay (Jiang et al.,2014b).The results were determined using a microplate reader (Spectra Max M2e,Sunnyvale,CA,USA) at 490 nm.
ROS evaluation
To assay the intracellular ROS production,HT22 cells were seeded on glass coverslip slides and lodged in 6-well plates.After appropriate stimulation with erastin,cells were loaded with 10 µM 2,7-dichlorodihydro-fluorescein diacetate for 10 minutes (CM-H2DCFDA,Thermo Fisher Scientific,Schwerte,Germany).Cell fluorescence was captured by the Axioskop microscope (Zeiss,Göttingen,Germany).Five microscopic fields for each experimental point were analyzed.Fluorescence intensity was processed by ImageJ analysis soft ware bundled with 64-bit Java 1.8.0_172 (ImageJ,National Institute of Health,Bethesda,MD,USA) and the results were expressed as a percentage of control.
Immunofluorescence
Formalin-fixed cells were incubated overnight at 4°C with primary anti bodies:GPX4 (rabbit,mAb;1:200,Cat# 52455,Cell Signaling,Frankfurt,Germany),cystine-glutamate antiporter (x-CT;rabbit mAb;1:200,Cat# 12691,Cell Signaling),FTH (rabbit,mAb;1:200,Cat# 4393 Cell Signaling),mtFt (rabbit,mAb,1:200,Cat# 31718,Abcam),NCOA4 (rabbit,mAb;1:200,Cat#66849,Cell Signaling),ACSL4 (rabbit,polyclonal,1:200,Cat# PA5-27137,Thermo Fisher Scientific).Briefly,Cells were pre-incubated for 1 hour at room temperature in PBS and 5% normal goat serum (Sigma),and primary antibodies were applied at 4°C for 24 hours in PBS plus 5% normal goat serum.Alexa Fluor 488 (Cat# A-32723) and Alexa 568 (Cat# A-11004) goat anti-mouse secondary antibodies (1:200,Molecular Probes,Göttingen,Germany).Adjacent wells that were not treated with the primary anti body were run for each well in parallel.After rinsing the cells with 0.1 M PBS,they were mounted in Dako fluorescent mounting medium (Dako,Hamburg,Germany).Fluorescent images were acquired using an Axio-Cam digital camera mounted on a Zeiss microscope (Carl Zeiss,Jena,Germany).
For semiquanti tative densitometric analyses of the immunoreactions,images were digitized with an Axiocam system (Zeiss,Göttingen,Germany;1030 ×1030 pixels,8-bit color depth),using NIH ImageJ soft ware (Image Processing and Analysis in Java,developer Wayne Rasband),as described previously(Rawashdeh et al.,2016).Six to eight ROIs were selected individually,and the relative optical density (ROD) of background staining was measured within the selected areas.
Evaluation of autophagy by assessment of fluorescent microtubuleassociated protein light chain 3 puncta
To confirm that erastin induces autophagosome formation,we examined and quantified the immunoreacti vity of light chain 3 (LC3)-II in HT22 neurons at 6,16,and 24 hours after ersatin treatment.The antibody directed against LC3-II was used at 1:200 dilution for 18 hours at 4°C (rabbit,mAb,Cat# 3868,Cell signaling).LC3,the microtubule-associated protein 1A light-chain 3,is normally located throughout the cytoplasm but becomes concentrated in autophagosomes during autophagy (Rami et al.,2017).The number of LC3 dots was determined by manual counting of fluorescent puncta in seven fields from three different slides with a 60× objective.The number of nuclei was evaluated by counting the number of 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei in the same field.The number of dots per cell was obtained by dividing the total number of dots by the number of nuclei in each microscopic field (Merck).
Western blotting
Cell homogenates were prepared in ten volumes of homogenization buffer (150 mM NaCl,20 mM Tris pH 7.5,1 mM ethylendiamintetraacetat,0.5% sodium deoxycholate,0.1% sodium dodecyl sulfate,1% Nonidet P-40),and polytron homogenization for 10 seconds.Aliquots were stored at -70°C and 30 µg of total protein was used per lane.Samples were resuspended to contain 30 µg of total protein in loading buffer,heated for 5 minutes at 95°C,and separated using a MINI-PROTEAN II electrophoresis system from Bio-Rad (Bio-Rad Laboratories,Feldkirchen,Germany) on either a 10% Tris-glycine gel,or a 15% Tris-glycine gel with 4% stacking gels.Gels were run at 180 V for 1 hour.The matched gel was soaked for 15 minutes in transfer buffer (25 mM Tris,190 mM glycine,20% methanol) and then transferred to a polyvinylidene difluoride membrane for 30 minutes at 100 mA constant current using a Bio-Rad Semi-Dry Blotting apparatus.Blots were blocked with 10% rehydrated nonfat dry milk (Sigma) for 1 hour at room temperature.Primary anti bodies against GPX4 (rabbit mAb;Cat#52455,Cell signaling),mtFt (rabbit pAb,Cat# 66111,Abcam,Berlin,Germany),FTH (rabbit mAb;Cat# #4393,Cell signaling),NCOA4 (rabbit mAb;Cat# 6684,Cell Signaling),x-CT (rabbit mAb;#12691,Cell signaling,Frankfurt,Germany),ACSL-4 (rabbit polyclonal,Cat# PA5-27137,ThermoFisherScientific),and β-acti n(Mouse mAb,Cat# A5316,Sigma) were used.
Western blots were performed with horseradish peroxidase-conjugated anti -rabbit IgG (Dako) using enhanced chemiluminescence Western blotting detection reagents (Amersham International,Sigma).To ensure equivalent protein loading,the stripped membranes were rehybridized with β-actin anti body as an internal control.The expression of protein in each group at 6,16,and 24 hours post-erastin incubation was examined by western blot assay.
To evaluate the results of the western blot assay,we scanned the film with an imaging densitometer (ChemiDoc XRS System,Bio-Rad,München,Germany)and quantified the optical density using a gel analysis software QuantiScan program from Biosoft (Bio-Rad Laboratories).Three blots from each group were examined.Rectangular regions were identified within the image and the background was substracted.The center of mass within the region and the net and gross total volume were calculated.The volume was calculated as the summed value of all pixels within the region.The optical intensity of target signals on a given western blot was normalized to the optical intensity of the β-actin signal on the same blot.The normalized signal intensities were then expressed as relative signal intensities (ROD %).
Stati stical analysis
Data are reported as the mean+SD.All assessments were performed by researchers blinded to treatments.Data were analyzed using GraphPad Prism 8.4.3 (GraphPad Soft ware,San Diego,CA,USA,www.graphpad.com).One-way analysis of variance with Tukey’spost hoctest was performed for experiments that involved more than two conditions.Significant differences between means at each time point were assessed by paired Student´st-test andP<0.05 was considered stati stically significant.
Results
Erastin-induced toxicity in HT-22 cells
Dose-response tests evaluated the effect of treatment of HT-22 neurons with different concentrations of erastin and showed decreased survival of neurons over time (MTT assay).Erastin decreased significantly the viability of hippocampal HT22 neurons.The cell survival rate was almost the lowest in HT-22 cells treated with 0.6 µM erastin for 12 hours.For our study,we selected the 0.5 µM erastin (Figure 1A).
Erastin-induced ROS production in HT22 cells
Another characteristic of ferroptosis is lipid peroxidation (Gao and Dixon,2016).To investigate whether erastin caused lipid peroxidation in the hippocampal HT22 neurons,we determined the levels of ROS.The ROS levels were significantly higher in the erastin group than in the control group (P<0.01).The analysis of fluorescent images clearly showed that erastin increased the amounts of ROS,confirming the oxidative role of erastin leading to neuronal toxicity (Figure 1B).
Erastin-induced changes in the levels of the autophagy related protein LC3-II in HT22 cells
The distribution of endogenous LC3-II in cells prior to and following erastin treatment was monitored by indirect immunofluorescence staining to examine whether autophagy was induced by erastin treatment.As demonstrated inFigure 2AandB,a greater number of dots were apparent in erastin-treated neurons compared with the control cells (6 and 16 hours:P<0.01;24 hours:P<0.05).
Neuroprotective effects of Fer-1,DFO,and 3-MA on HT22 cells
Fer-1 or DFO were preadministered to determine if ferroptosis was involved in erastin-induced toxicity.The results indicated that Fer-1 or DFO significantly reduced the cell death induced by erastin (P<0.01;Figure 3AandB).In addition,3-MA as an inhibitor of autophagy was used to determine if autophagy was involved in erastin-induced ferroptosis.The autophagy inhibitor 3-MA protected hippocampal HT22 neurons from ferroptotic cell death induced by erastin (Figure 3C).In addition,we found that 3-MA attenuated the ROS levels in erastin-treated cells (P<0.05;Figure 3C).
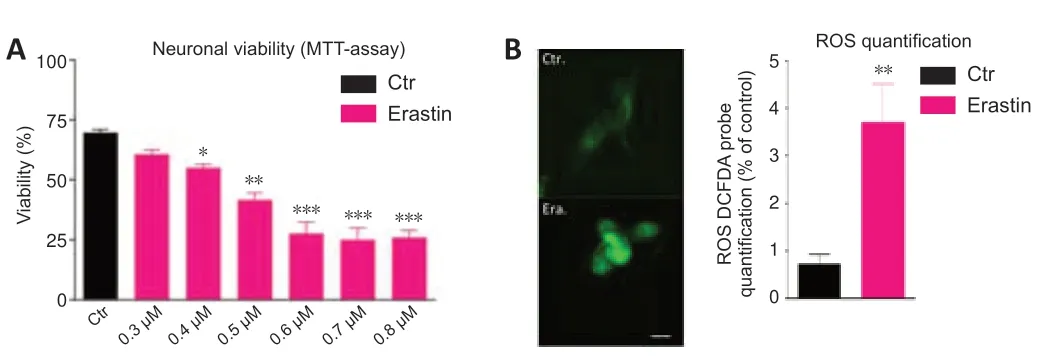
Figure 1|Erastin induces cell death (A) and ROS production (B) in hippocampal HT22 neurons.
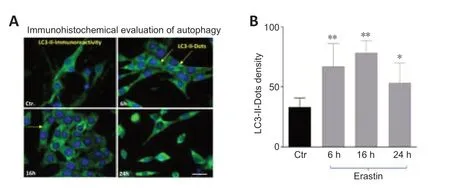
Figure 2|Erastin induced changes in the levels of the autophagy related protein LC3-II.
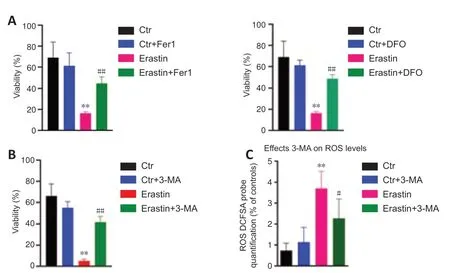
Figure 3|Effects of DFO/Fer-1 and 3-MA on ersati n-induced ferroptosis.
Erastin-reduced GPX4 and x-CT-expression in HT22 cells
As biomarkers of ferroptosis,the expression of x-CT (a sodium-independent cystine-glutamate anti porter) and GPX4 were analyzed.We determined the expression of GPX4 after treating the HT22 cells with the GPX4 inhibitor and ferroptosis inducer,erastin.The densitometric analysis of GPX4-immunoreacti vity and GPX4-immunoblots revealed a stati stically significant decrease upon erastin treatment as early as 6 hours post-incubation (6 hours:P<0.05;16 and 24 hours:P<0.01;Figures 4and5B).The x-CTimmunoreactivity as well as the density of immunoblots was more intense in controls than in erastin-treated cells.The x-CT levels were significantly decreased at all the time points examined in erastin-treated cells (P<0.01;Figures 4and5A).
Erastin does not alter significantly the FTH and mtFt levels in HT22 cells
We determined the expression levels of FTH and mtFt after treating the HT22 cells with the GPX4 inhibitor and ferroptosis inducer,erastin.FTH and mtFt levels were measured in control and erastin-treated cultures at 6,16,and 24 hours post-incubation.Western blot and immunofluorescence analysis revealed no remarkable alterations in FTH/mtFt expression upon erastin treatment (P>0.05;Figures 4and5CandD),except for a slight transient increase in FTH levels at 6 hours after incubation with erastin.
Erastin-induced alterations in NCOA4-and ACSL-4-levels in HT22 cells
NCOA4 was identified as a key player in ferritinophagy,serving as an autophagic cargo receptor to facilitate the delivery of ferritin for lysosomal degradation (Mancias et al.,2014).Immunofluorescence and western blot analysis showed a gradual decrease in NCOA4 levels at 6,16,and 24 hours post-incubation with erastin (Figures 4and5E).
ACSL4 is one of the main enzymes which promote ferroptosis (Yuan et al.,2016).Cell lines with deleted ACSL4 gene showed less susceptibility to ferroptosis inducers such as erastin and 1S,3R-Methyl 2-(2-chloroacetyl)-2,3,4,9-tetrahydro-1-[4-(methoxycarbonyl)phenyl]-1H-pyrido[3,4-b]indole-3-carboxylat (RSL-3) (Dixon et al.,2015;Doll et al.,2017).In addition,in some cell lines,the abundance of ACSL4 could be regarded as a biomarker of ferroptosis sensiti vity (Yuan et al.,2016).Immunofluorescence and western blot analysis showed a gradual decrease in ACSL4 levels at 6,16,and 24 hours post-incubation (Figures 4and5F).
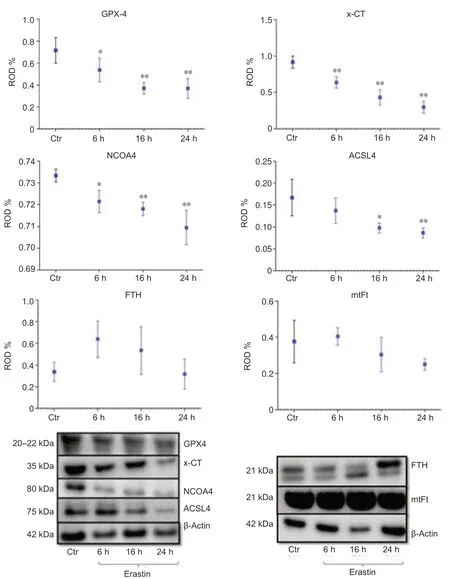
Figure 4|Western blot assay and densitometric analysis of immunoblots of GPX4,x-CT protein,NCOA4,ACSL4,FTH and,mtFt 6,16 and 24 hours after incubation with 0.5 μM erastin in HT22 neurons.
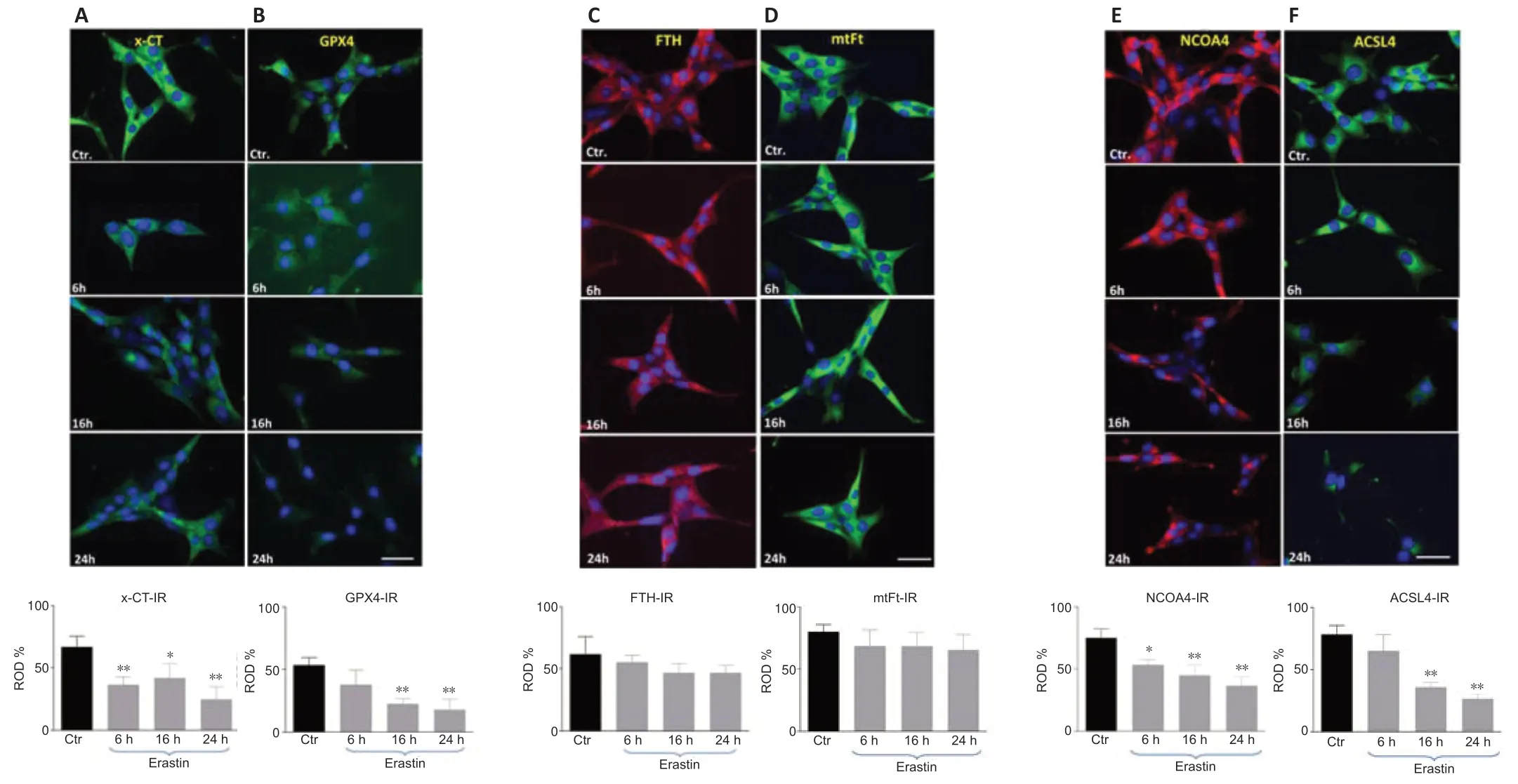
Figure 5|The x-CT-(A),GPX4 (B),FTH (C),mtFt (D),NCOA4 (E) and ACSL4 (F) immunoreacti vities (IR) in HT 22 neurons collected from controls and at 6,16,and 24 hours after incubation with erastin.
Discussion
Although neuronal death seems to be a multi factorial process,there are now indications that neuronal iron overload and release of stored intracellular iron are critical events leading to irreversible neuronal damage.Iron homeostasis is indispensable for neuronal cell function,however,deregulation of cellular iron is potenti ally harmful due to its reactive nature (Arosio and Levi,2002;Kruer,2013;Dixon and Stockwell,2014;Bodgan et al.,2016).Deregulation of cellular iron leads to increased ROS.Increased ROS leads in turn to impairment of the cellular antioxidant system,which may exacerbate the suscepti bility of neurons to iron.Under stress conditions,cells must actively buffer their intracellular iron using cytosolic iron storage proteins such as ferritin (Arosio and Levi,2002;Arosio et al.,2017).Our initial goal was to determine whether there were alterations in the expression levels of ferroptosis key players in HT22 neurons following erastin treatment.We first addressed GPX4-protein,which is a strong antioxidant enzyme (Dixon et al.,2012;Gao et al.,2016).Accordingly,we demonstrated that erastin treatment induced a significant ROS accumulation and dramatically downregulated the GPX-4 expression in HT22 neurons.This supports the hypothesis that the cell death induced by erastin in HT22 neurons is ferroptotic and indicates that neurons are specifically sensitive to GPX4 depletion.In this context,it is known that GPX4-deletion triggers degeneration of spinal motor neurons (Chen et al.,2015) and that Gpx4-ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in the brain (Yoo et al.,2012).As matter of fact,GPX4-knockout mice exhibit a selective degeneration of CA1 pyramidal hippocampal neurons and interneurons (Seiler et al.,2008;Hambright et al.,2018;Ingold et al.,2018).
The x-CT levels were significantly reduced in HT22 neurons after erastin treatment.In general,a decrease in x-CT levels should stabilize glutamate homeostasis and prevent neuronal damage.However,it is not the case in our system,cell damage occurred even with a decrease in x-CT levels.This lets us assume that the capacity of this process seems to be limited and not powerful enough to rescue neurons from the damaging effects of erastin.
Seeing that ACSL4 expression is well correlated with ferroptosis sensiti vity in various cancer cells and is considered a biomarker of apoptosis (Yuan et al.,2016;Arosio et al.,2017),this is not the case in hippocampal HT22 neurons.In our study,erastin reduced the ACSL4 levels in hippocampal HT22 neurons.Comparing HT22 neurons with cancer cell lines,the reduction of ACSL4 levels in erastin-treated HT22 cells should mean consequently an attenuation of ferroptosis.On the contrary,we found in our study that ferroptosis occurred despite reduced ACSL4 levels in HT22 neurons.We can assume that neuronal and cancer cells may respond differently to ferroptosis inducers.
Elevated cytosolic iron levels triggered the expression of neuronal ferritin,presumably to bind and buffer iron.It is likely that ferritin is one of the metalloproteins which play an active,protective role within neuronal cells by diminishing the availability of reactive iron upon accumulation,and readily providing iron for utilization under physiological conditions.The ferritin homeostasis is checked by NCOA4,which regulates the lysosomal degradation of ferritin via a selective form of autophagy called ferritinophagy (Hou et al.,2016;Gryzik et al.,2017;Hassannia et al.,2018).Accordingly,we next determined whether a change in NCOA4 expression occurs during erastininduced ferroptosis in HT22 neurons.We observed that the protein levels of NCOA4 were remarkably reduced following HT22 neurons were treated with erastin.Compared with cancer cells,the decrease of NCOA4 levels in erastin-treated HT22 cells,should mean consequently a reduction of NCOA4-mediated ferritinophagy in HT22 cells and consequently an attenuation of ferroptosis.This is not the case in our model of erastin-induced ferroptosis in HT22 cells.Ferroptosis occurred despite reduced NCOA4 levels.These discordant results may be a consequence of a number of differences between tumor-derived cell lines and neurons,including the relative dependence on NCOA4-mediated ferritinophagy for iron balance in cell culture models and differences in iron availability between cells.However,our results are in accordance with the studies of Gao and Dixon (2016),who have shown that erastin reduced NCOA4-levels in mouse embryonic fibroblasts,and this was accompanied by NCOA4 autophagic degradation.
Ferriti nophagy is the process of autophagic degradation of the iron storage protein ferriti n,which is critical for the regulation of cellular iron levels (Tang et al.,2018).Accordingly,we next determined whether a change in ferritin expression (FTH and mtFt) occurs during erastin-induced ferroptosis in HT22 neurons.FTH/mtFt levels were not remarkably altered in erastin-treated HT22 cells,which may indicate that ferriti nophagy does not occur or it is of secondary importance in this experimental model.In contrast,recent data support the protective effect of ferritin accumulation,because the lack or deficiency of mitochondrial ferritin exacerbates neurological deficits and oxidative stress in a traumatic brain injury model (Wang et al.,2017,2018).In addition,it has been shown that FTH upregulation in neuroblastoma was an important neuroprotective response to the oxidative insult caused by the neurotoxic agent rotenone (Mackenzie et al.,2008).A protective role of ferritin was also observed in dopamine neurons of the substanti a nigra after treatment with the proteasome inhibitor lactacystin (Zhu et al.,2007).
To further confirm the eventual involvement of autophagy in ferroptosis,we first,assessed by indirect immunofluorescence the density of autophagosomes in erastin-treated HT22 cells,and second,we tested the effects of the autophagy inhibitor,3-MA on neuronal viability after erastin treatment.We found indeed that autophagy was enhanced under erastintreatment and that 3-MA attenuated ROS production and ameliorates significantly the cell viability in erastin-treated hippocampal HT22 neurons.Consistently,inhibition of autophagy in erastin-treated hippocampal neurons abrogated ferroptotic cell death.Therefore,ferroptosis seems to share some features of the autophagic cell death process.
It has been reported that NCOA4 levels were enhanced in multiple cancer cell lines and that NCOA4-dependent ferriti nophagy can promote ferroptosis by releasing free iron from ferritin (Ingold et al.,2018).However,to date,there is no direct evidence associating ferriti nophagy and ferroptotic cell death in neurons.
In summary,the present study provides important evidence that hippocampal HT22 neurons respond differently from cancer cell lines to the ferroptosis inducer erastin.In addition,autophagy seems to promote erastin-induced ferroptosis.There are some limitations to the present study.For instance,we did not investigate all downstream regulatory mechanisms of NCOA4-ferritinophagy in erastin-treated cells.Besides ferritinophagy,a further understanding of the functional characterization of different pathways leading to ferroptosis,NCOA4-degradation,and its contribution to intracellular and systemic iron homeostasis,will be important.
Acknowledgments:Immortalized mouse hippocampal cell line (HT22) was a gift from Prof.David Schubert (Salk Institute,La Jolla,CA,USA).
Author contributions:NH and AR contributed to the study conception and design,material preparation,data collection,and analysis.AR wrote the paper.Both authors read and approved the final manuscript.
Conflicts of interest:The authors have no relevant financial or nonfinancial interests to disclose.
Open access statement:This is an open access journal,andarticles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Bystanders or not? Microglia and lymphocytes in aging and stroke
- Alzheimer’s disease risk after COVID-19: a view from the perspective of the infecti ous hypothesis of neurodegeneration
- Serine and arginine rich splicing factor 1: a potenti al target for neuroprotection and other diseases
- Can glial cells save neurons in epilepsy?
- Lights for epilepsy: can photobiomodulation reduce seizures and offer neuroprotection?
- The landscape of cognitive impairment in superoxide dismutase 1-amyotrophic lateral sclerosis