
Alzheimer’s disease risk after COVID-19: a view from the perspective of the infecti ous hypothesis of neurodegeneration
2023-02-24 05:24EugeniaOliveraAlbanyezLilaCarnigliaCarlaCarusoMercedesLasagaDanielaDurand
中国神经再生研究(英文版) 2023年7期
Eugenia Olivera,Albany Sáez,Lila Carniglia,Carla Caruso,Mercedes Lasaga,Daniela Durand
Abstract In light of the rising evidence of the association between viral and bacterial infections and neurodegeneration,we aimed at revisiting the infectious hypothesis of Alzheimer’s disease and analyzing the possible implications of COVID-19 neurological sequelae in long-term neurodegeneration.We wondered how SARS-CoV-2 could be related to the amyloid-β cascade and how it could lead to the pathological hallmarks of the disease.We also predict a paradigm change in clinical medicine,which now has a great opportunity to conduct prospective surveillance of cognitive sequelae and progression to dementia in people who suffered severe infections together with other risk factors for Alzheimer’s disease.
Key Words:Alzheimer;amyloid beta;antimicrobial;cognitive decline;COVID-19;infectious hypothesis;long-term sequelae;neurodegeneration;neuroinflammation;neurological symptoms;neurotropism;SARS-CoV-2
Introduction
It is now vox populi that coronavirus disease 2019 (COVID-19) leaves real neurological sequelae,including loss of taste and smell,or even encephalitis,cerebrovascular disorders,or long-term cognitive deficits.This is an issue of major concern worldwide.How long can these signs persist? Is there an actual risk of long-term brain damage or a real increased risk of developing some kind of dementia over time? Among the multiple existing voices,information may not always be reliable or scientifically supported.In this context,revisiting the infectious hypothesis of Alzheimer’s disease (AD)gains major relevance.Under this hypothesis,the root cause of AD is a pathogen that triggers an immune reaction involving amyloid-β (Aβ) as an anti microbial agent.Thereafter,some kind of disequilibrium in Aβ turnover(probably related to the aging process) leads to its pathological accumulation and AD development.The present article aimed at reviewing the most recent literature supporting the infecti ous hypothesis of AD,and addressed the probable risk of developing Alzheimer-like dementi a after severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection,based on evidence linking viral infections and amyloidosis.
Data Sources
The following terms and combinations of terms were searched in PubMed(https://pubmed.ncbi.nlm.nih.gov/) and Scholar Google (https://scholar.google.com/) databases or in https://clinicaltrials.gov/: “COVID-19”,“SARSCoV-2”,“infection”,“neurological sequelae”,“dementia”,“Alzheimer”,“amyloidosis”,“Infectious Hypothesis of Alzheimer” “ACE2”,“cognitive deterioration”,“prospective”,“retrospective”,“excitotoxicity”,“glutamate receptors”.Additional disease-specific parameters were “herpes simplex type 1” OR “gingivitis” PLUS “neurological disorders” OR “Alzheimer”.Preprints were excluded.Selected articles were also searched for references.All articles referring to psychiatric conditions (including AD) predisposing to COVID-19 infection were excluded.An additional filter for articles published in 2022 was included to focus on clinical studies with a wider observational window.Detractor positions to the Infecti ous Hypothesis of AD eti ology were specifically searched for to cover the two currents.Date of the last search:May 31,2022.
Infecti ous Hypothesis: Its Origins
Microbes are present in most elderly brains (Link,2021).Spirochaeteinduced syphilitic dementi a formerly became popular through famous arti sts of the 18thcentury.Moreover,the occurrence of “resilient” brains showing widespread Aβ deposition and tangles in absence of signs of either dementi a or neuroinflammation emphasizes the role of aberrant innate immunity in AD(Moir et al.,2018).
The infecti ous hypothesis proposes a pathogen as the root cause of sporadic AD and is based on increasing evidence of some viruses and bacteria frequently found in histopathological analysis of post-mortem brain from AD patients.The first approach to an infection as the possible eti ology of AD was postulated by Dr.Oskar Fischer,contemporaneously with Alzheimer’s first report in 1907 (Fischer,1907).Since then,several scienti sts have investigated the link between pathogens and AD development.In the beginning,infecti ous hypothesis followers were ignored or ridiculed,especially because the proposition was seen as a replacement for the Amyloid Cascade Hypothesis.However,over the last decades,the hypothesis has gained relevance and adepts among neuroscientists,and valuable clues regarding how both theories can coexist have been found (Abbott,2020).Since the 1990 decade,different laboratories have associated infection with the etiology of AD.Spirochetes andC.pneumoniaewere found in blood,cerebrospinal fluid(CSF),and brain tissue from AD individuals (Miklossy,1993;Balin et al.,1998).Systemic infection byC.pneumoniaewas associated with a 5-fold increase in AD occurrence (Balin et al.,2008).An association study by Bu et al.(2015)including 128 AD patients and 135 healthy controls provides evidence of infectious burden,involving viruses and bacteria,which is associated with AD.Sepsis survivors were associated with new cognitive impairment and functional disability (Iwashyna et al.,2010).Moreover,HIV-associated neurological disorders cause AD-like symptomatology (Smail and Brew,2018).These studies support the notion that AD might have an infecti ous eti ology.Although no consensus exists on how an infectious pathogen can explain the pathological features of AD,the most accepted view holds that proinflammatory cytokines produced by innate immunity during an infection (tumor necrosis factor-α,interleukin-1β,interferon-γ) promote an inflammatory process,consequently increasing Aβ synthesis and accumulation,as well as hyperphosphorylation and aggregation of tau(Seaks and Wilcock,2020).Based on genome-wide association study analysis of polymorphisms in AD individuals,Porcellini et al.(2010) argue that the concomitant presence of several polymorphisms in genes related to suscepti bility to brain viral infections in the same individual (NC-2 or herpes virus entrance-B,apolipoprotein E (APOE4),complement receptor CR1,among other genes) might represent a genetic signature of AD.On the same line,Readhead et al.(2018) found genes involved in virus response differenti ally expressed between healthy controls and preclinical AD individuals.
Although substanti al evidence supports the infecti ous hypothesis as a possible origin of AD,like any paradigm-changing theory,it is sti ll regarded with some skepticism.The main arguments emphasize the need for large longitudinal population data sets to corroborate the relevance and reproducibility of results.Further,John Hardy affirmed,concerning the article of Readhead et al.(2018): “My concern about this work is that I find it difficult to square with the occurrence of AD in all Down’s syndrome and in all carriers of some APP and PSEN mutations […] We have to suppose that Down’s cases and the mutation cases either have a different disease mechanism or that for some reason,they are uniquely susceptible to these infections” (https://www.alzforum.org/news/research-news/aberrant-networks-alzheimers-ti edherpes-viruses).Despite these public declarations,we were unable to find any published articles arguing against the infectious hypothesis.Mawanda and Wallace (2013) provided some valuable criticisms of the findings in this field,but stated that “the amount of evidence suggestive of an association between AD and an infecti ous cause is too substanti al to ignore”.They mainly argued that no single infecti ous agent was shown to be causative of AD;that several studies used indirect methods for pathogen detection;or that,alternatively,AD neuropathology might increase the susceptibility of affected areas to infections.Anyway,the same pioneers of the infecti ous hypothesis are also cauti ous about referring to pathogens as a direct cause of AD,but instead as a prequel of the Amyloid Hypothesis.A schematic image of the main points of this hypothesis is shown inFigure 1.
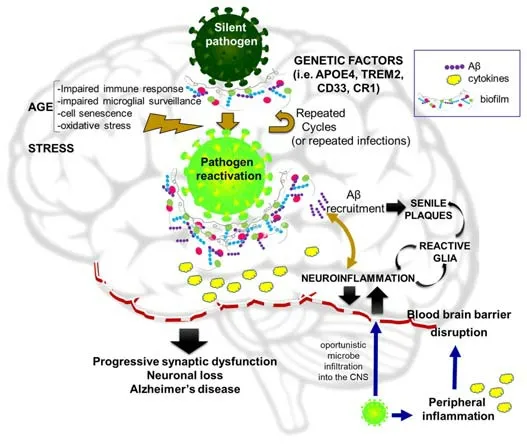
Figure 1|The infecti ous hypothesis of Alzheimer’s disease eti ology.
Infections and Neurodegenerative Diseases
Sipilä et al.(2021) studied the risk of AD and other dementi as across a wide range of hospital-treated bacterial and viral infections in two large cohorts(260,490 people in the primary cohort and 485,708 people in the replication cohort) with long follow-up periods.Hospitalization for any infecti ous disease was associated with increased dementi a risk even after restricting the analysis to new dementi a cases,the risk was higher in the cases of repeated episodes of hospital-treated infections.Among types of infections,central nervous system (CNS) and extra-CNS infections were both significant risk factors for dementi a,whereas,among types of dementi a,vascular dementi a correlated better with infections than AD did.Based on the absence of infection specificity,the authors suggested that “increased dementi a risk is driven by general inflammation rather than specific microbes”.Lövheim et al.(2018)also stated that the interaction between two different viral infections,but not a single virus,was associated with AD development.
Herpes simplex virus type 1 (HSV-1) can live within the host establishing a latent infection,thus evading the immune response.It can be then reactivated to a lytic state,triggered by stress or co-infections that weaken the immune system (Jamieson et al.,1991).HSV-1 can enter the CNS via axonal retrograde transport to infect the cell nucleus of sensory ganglia(Shimeld et al.,2001).Herpes simplex encephalitis can damage the limbic system,a region particularly affected by AD (Roos,2014).A hazard ratio of 2.564 for the development of dementi a in the HSV-infected cohort relative to the age-matched non-HSV cohort was found.It is known that carrying the APOE4 gene variant is a risk factor for AD.APOE and HSV-1 use the same binding site to enter the cell,the heparan sulfate proteoglycans.Linard et al.(2020) showed that APOE4 carriers with frequent HSV reactivation are at higher risk of AD and,among APOE4 non-carriers,no association was found between HSV-1 and AD.This may be consistent with murine studies in which APOE4 was associated with a higher HSV-1 load in mouse brains and an increased risk of cold sores compared to APOE3.Remarkably,a reduction of dementia development risk in patients affected by HSV infections was found upon treatment with antiherpetic medications.A Phase II trial is currently evaluating the efficacy of valacyclovir in slowing cognitive decline in 130 people with mild AD who tested positive for HSV-1 or -2 (ClinicalTrials.gov Identifier: NCT03282916).However,other groups did not detect HSV in elderly brains or an association with AD risk factors.By using complementary,very sensitive methods of pathogen sequence detection from three independent repositories,Allnutt et al.(2020) concluded that there are no significant differences in viral detection between AD and non-AD controls.A discrepancy is also observed when determining anti-HSV antibodies in a longitudinal population-based cohort of elderly subjects followed for 14 years (Letenneur et al.,2008).Controlled for age,gender,educational level,and APOE4 status,IgM-positive subjects showed a significantly higher risk of developing AD while no significantly increased risk was observed in IgGpositive subjects.
Periodonti ti s is a generalized infection of the supporting tissues of teeth and can be caused by the progressive accumulation of bacterial biofilm made of oral pathogens such asP.gingivalisandA.actinomycetemcomitans(Cheng et al.,2016).Every time a person infected with periodonti ti s brushes his/her teeth,bacteremia risk increases.Successive bacteremia events provoke the release of pro-inflammatory virulence factors that,over time,weaken the blood-brain barrier (BBB) and allow pathogens to enter the CNS (Olsen,2021).P.gingivaliscan modify the peripheral and intracerebral immune responses and therefore lead to the progression of neurodegenerative diseases such as AD.Ilievski et al.(2018) and Zeng et al.(2021) found that at least two bacterial enzymes (cathepsin B and gingipains) can interact with the amyloid precursor protein (APP) and neuronal tau,resulting in the formation of Aβ and neurofibrillary tangles.Virulence factors ofP.gingivaliscan contribute to AD and clinical studies are currently evaluating the beneficial effect of gingipain inhibitors in AD patients (Dominy et al.,2019).The chronic nature of lowlevel infections such as gingivitis and periodonti ti s could affect the capacity of defense by the microglial cells causing Aβ to accumulate into plaques(Olsen and Singhrao,2020).A previous study has shown that patients with periodontal disease for at least 10 years have double the risk of developing AD (Chen et al.,2017).
Far from being an exhaustive review addressing bacterial or viral pathogens linked to neurodegenerative disorders,the reported evidence serves as a framework to introduce the possible association of SARS-CoV-2 with AD.Whether this implies a direct causal relationship or derives from a chronic inflammatory response to the pathogen remains to be determined,but inflammation is a very significant component of the disease etiology.Complement factors and clustering of activated microglia within plaques,as well as gliosis,are well documented in AD brains,whereas genes encoding inflammation-related proteins like clusterin and complement receptor CR1 are among AD risk factors identified by genome-wide association studies(Eikelenboom et al.,2012).A 17-year follow-up study by Tao et al.(2018)including longitudinal C-reactive protein measurements showed that APOE4 coupled with chronic systemic low-grade inflammation was associated with increased risk and accelerated onset of AD in a pattern dependent on the C-reactive protein level.The accumulative detrimental effect of low-grade chronic inflammation can be enhanced by the expression of high-risk AD variants of genes like APOE,TREM2,CR1,or CD33,which are involved in innate immunity (Bocharova et al.,2021).
Pathogens and Amyloidosis: the Mechanistic Link to the Eti ology of Alzheimer’s Disease?
The infectious hypothesis was quickly accompanied by the complementary Antimicrobial Protection Hypothesis conceived by Moir and Tanzi’s group.These researchers highlight the physiological role of Aβ as “an ancient,highly conserved effector molecule of innate immunity with anti microbial properti es”(Moir et al.,2018).Within this conceptual framework,senile plaques should be seen as a manifestation of the disease,not the cause,becoming the“friendly fire” contributing to neurotoxicity.Thus,the anti microbial protection hypothesis of AD does not necessarily invalidate the amyloid cascade hypothesis.The seminal work demonstrating that Aβ is an antimicrobial pepti de compared Aβ to LL37 (a powerful anti microbial agent active against various bacteria and fungi) and showed that synthetic Aβ reduced the growth of pathogens likeE.faecalis,S.aureus,andC.albicans,among others by up to 200-foldin vitro(Soscia et al.,2010).Aβ also had anti viral acti vity against HSV-1 (Bourgade et al.,2016) and influenza A (White et al.,2014).The presence of microbes serves as a surface for nucleation of amyloid aggregates,thereby allowing for amyloid deposition as a biofilm matrix (Fulop et al.,2018;Figure 1).Direct interaction between βAPP and HSV-1 was reported by Cheng et al.(2011) and acyclovir was able to block HSV1-induced Aβ and tau pathology(Wozniak et al.,2011).Aβ oligomers inhibited HSV-1 infectionin vitroand protected mice from acute viral encephalitis induced by HSV-1 (Eimer et al.,2018).AD brain homogenates from Aβ-enriched regions showed elevated antimicrobial activity againstC.albicanscompared to non-AD samples(Soscia et al.,2010).In agreement with these results,elevated infection rates were reported as a side effect of a clinical trial using the Aβ-lowering drug tarenflurbil (Green et al.,2009).However,there are also counterarguments to this hypothesis,such as the study by Bocharova et al.(2021),which found that the 5×FAD genotype failed to protect mice against HSV-1 infection.They showed that young 5×FAD mice that survived infection cleared HSV-1 without triggering Aβ aggregation;whereas,in aged mice,HSV-1 replication was inhibited in Aβ-positive areas but no evidence of viral entrapment by Aβ was found.Instead,the protective effect may be attributable to chronic microglial activation.
Neurological Manifestations of SARS-CoV-2
Despite literature reporting that the hazard ratio for SARS-CoV-2 infection is higher in people with AD and other neurodegenerative disorders,the goal of this review was to analyze evidence of the risk of developing AD after COVID-19 infection;therefore,our search was narrowed in this direction.At this point,we should stress that all evidence reported here was used to infer,under the infecti ous hypothesis view,whether there is a scientificallysupported potential risk of developing AD among SARS-CoV-2 infected subjects.However,any approach to this question should consider the undeniable effects of lockdown,isolation,and isolation-linked mood disorders on brain function,or even the social and economic impact of the pandemic on the global health of people.
In addition to respiratory and cardiac manifestations,one-third of patients with COVID-19 developed neurological symptoms like headache,disturbed consciousness,paresthesia,brain tissue edema,stroke,neuronal degeneration,or encephalitis (Lou et al.,2021).Even patients without observable neurological manifestations exhibited neurological microstructure changes at the 3-month follow-up (Qin et al.,2021).Pistarini et al.(2021)and Mazza et al.(2021) found a high prevalence of cognitive impairments,especially executive functions and motor coordination,in both COVID-19 and post-COVID-19 patients.Fleischer et al.(2021) carried out a prospective,cross-sectional study of 102 SARS-CoV-2 PCR-positive patients.Of these,59.8% of patients had neurological involvement including general weakness and cognitive decline or delirium (24.5%),impaired taste or smell (9.8%),or severe events including cerebral ischemia (23.5%).In a longitudinal study (Atahualpa project by Del Brutto et al.,2021),cognitive evaluations in previously cognitively healthy individuals at 6 months after the SARSCoV-2 outbreak yielded an odds ratio for developing cognitive decline of 18.1 times higher among SARS-CoV-2 seropositive individuals.Taquet et al.(2021) carried out a retrospective cohort study using the electronic health record network (with over 81 million patients analyzed) and estimated the incidence of 14 neurological and psychiatric outcomes in the 6 months after a confirmed diagnosis of COVID-19: intracranial hemorrhage;ischemic stroke;parkinsonism;Guillain-Barré syndrome;nerve,nerve root,and plexus disorders;myoneural junction and muscle disease;encephalitis;dementia;psychotic,mood,and anxiety disorders;substance use disorder;and insomnia.Among 236,379 patients diagnosed with COVID-19,the esti mated incidence of a neurological or psychiatric diagnosis in the following 6 months was 33.62%,with 12.84% receiving their first such diagnosis,the incidences being higher among patients who had been admitted to an intensive care unit.The values were significantly higher for patients who had COVID than those who had influenza or other respiratory tract infections as control cohorts.By contrast,in another cohort (Mattioli et al.,2021),cognitive impairments detected 4 months after mild-moderate SARS-CoV-2 infection were not significantly different from non-COVID-19 cases and Kanberg et al.(2021) found a normalization of CNS injury blood biomarkers after 6 months of COVID diagnosis,regardless of previous disease severity or persisting neurological symptoms.
We next restricted the search to papers published only in 2022 to include longitudinal,prospective,or retrospective studies with a wider time window for observational changes.According to data from COVID patients symptomatic after 2 months from diagnosis,85% still reported symptoms one year after onset,with some symptoms showing decreased prevalence over time (e.g.,loss of taste/smell,brain fog/attention deficits),others a stable prevalence over time (e.g.,dyspnea),and others increased prevalence over time (e.g.,paresthesia) (Tran et al.,2022).This study reported a slightly decreased prevalence of memory problems one year after the onset (55.7%to 50.2%).A retrospective cohort study by Kim et al.(2022) of predictors for new-onset mental disorders among patients with mild to moderate COVID-19 showed that 22.3% of patients were newly diagnosed with mental disorders during hospitalization,with length of stay and self-reported depressive symptoms at the time of admission being among the main risk factors for psychiatric diagnosis.Impaired Mini-Mental State Examination performances were highly prevalent in mild-to-moderate COVID-19 patients (26.3%),but,surprisingly,intensive care unit-admitted patients made fewer errors on the Mini-Mental State Examination than those not admitted,after adjusting for risk factors and age (Manera et al.,2022).
In addition,brain changes including glucose hypometabolism and neurotransmitter alterations detected by positron emission tomography in COVID patients have been reviewed by Fontana et al.(2020).A particular longitudinal study by Douaud et al.(2022) investigated brain changes in participants of UK Biobank who were imaged twice by magnetic resonance imaging,before and after COVID-19,a design that made it possible to discard any previous alteration which might lead to misinterpretation of disease effects.The authors identified a reduction in grey matter thickness and tissue contrast in the orbitofrontal cortex and parahippocampal gyrus (a limbic region that has a crucial,integrative role for episodic memory);changes in markers of tissue damage in regions that are functionally connected to the primary olfactory cortex;and reduced global brain size in the SARSCoV-2 cases compared to controls.These changes correlated with a greater cognitive decline between the two time points and did not overlap with those presented in non-COVID-19 pneumonia cases.Table 1summarizes data from these studies.
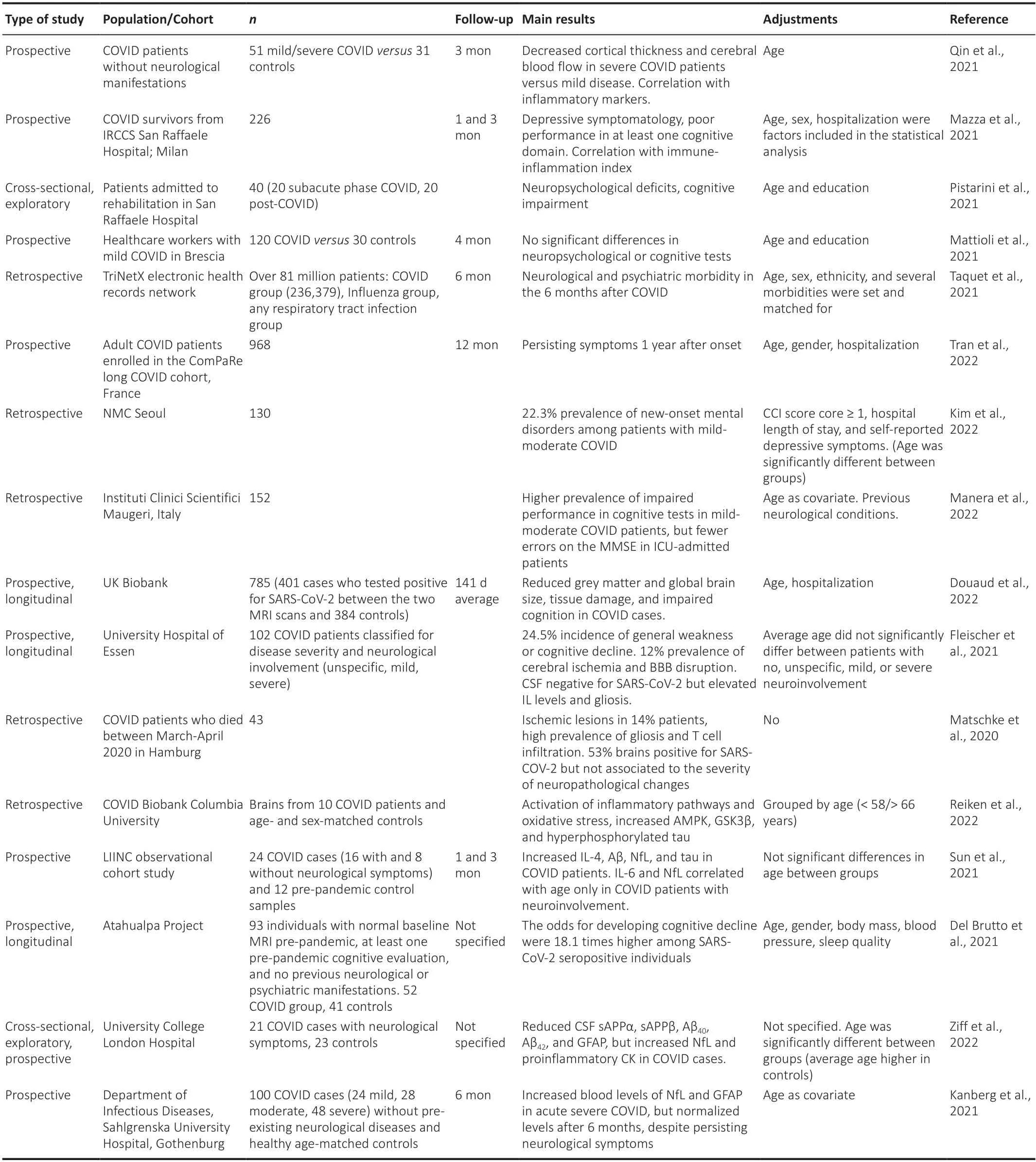
Table 1|Summary of research studies investigating neurologic involvement in COVID-19
To sum up,the real long-term cognitive alterations in post-COVID patients,as well as the causal link with SARS-CoV-2 infection,are sti ll elusive.However,considering the slow progression of AD since the first brain changes,cognitive performance should be followed up over a much longer period.
On the other hand,after the coronavirus pandemics in 2002 and 2012,one in five recovered individuals reported depressed mood,insomnia,anxiety,irritability,fatigue,and sleep disorders (de Erausquin et al.,2021).Then,is there a common mechanism for coronaviruses leading to neurological symptoms? Are these mood disorders natural after any overwhelming disease? Or do these similarities between diseases support the multiple pathogen cause of “infecti ous” AD?
SARS-CoV-2 Neurotropism versus Systemic Inflammation
Beta-coronaviruses including SARS-CoV-2 are thought to invade the CNS by binding their spike proteins to the angiotensin-converting enzyme 2 (ACE2)receptors on the olfactory epithelium.Compared to other CoVs,SARS-CoV-2 spike protein displays a 10–20-fold increased affinity toward ACE2 (Wrapp et al.,2020).The first evidence for neurotropism of the virus relied on the first neurological symptom described for COVID-19,anosmia.Interestingly,hyposmia has also been reported in early-stage AD (Meinhardt et al.,2021).Human CoV infection,along with other respiratory virus infections such as influenza,is shown to spread throughout the CNS,especially the temporal region and hippocampus associated with learning/cognitive changes(Dolatshahi et al.,2021).Besides ACE2 receptors,viral transmission is supposed to include trans-synaptic transfer across infected neurons,involving the olfactory nerve,as reviewed in (de Erausquin et al.,2021).The olfactory tract connects to brain regions closely related to cognition and memory.The presence of SARS-CoV-2 in the olfactory bulb leads to the activation of nonneuronal cells,such as mast cells,microglia,and astrocytes,and the release of pro-inflammatory cytokines (Ciaccio et al.,2021).The brain damage might also be a consequence of the loss of sensory input due to anosmia (Douaud et al.,2022).Furthermore,neurons and glia also express ACE2 and can internalize SARS-CoV-2.Neuronal infection is achieved in mice humanized at the ACE2 gene (Song et al.,2021).In turn,the occupation of ACE2 with the virus may lead to a decline in ACE2 acti vity,which acts as a neuroprotective factor (Dolatshahi et al.,2021).
In addition to ACE2,SARS-CoV-2 can enter the brain via other molecular mediators like neuropilin-1,transmembrane serine protease,CD-147,cathepsin L,and two pore segment channel-2.In contrast to ACE2,CD147 is greatly expressed in the brain (Qiao et al.,2020).Neuropilin-1 is not only expressed in the respiratory epithelium but also the olfactory epithelium,and is highly expressed in endothelial cells,excitatory neurons,and nasal cavity epithelial cells (Iadecola et al.,2020),which may mediate virus entry into the CNS.In COVID-19 postmortem brains,expression of ACE2 was highest in oligodendrocytes,while transmembrane serine protease 2 and transmembrane serine protease 4 were highest in neurons,cathepsin L was highest in microglia,and two pore segment channel-2 was highest in astrocytes (Matschke et al.,2020).
However,direct evidence of SARS-CoV-2 neurotropism is more elusive.Viral components have been detected in brains or CSF (Hosseini et al.,2020;Huang et al.,2020;Khodamoradi et al.,2020;Matschke et al.,2020;Moriguchi et al.,2020;Xiang et al.,2021).Paniz-Mondolfi et al.(2020) reported the postmortem presence of the virus in neural and capillary endothelial cells in the frontal lobe tissue of a COVID-19 patient,with clinical correlates of worsening neurologic symptoms.In contrast,neither viral RNA nor particles were found in brain tissue by other researchers (Helms et al.,2020;Edén et al.,2021;Pilotto et al.,2021).Gagliardi et al.(2021) detected viral RNA in the frontal cortex only by using a very sensitive method (droplet digital PCR),suggesting that SARS-CoV-2 does not actively infect and replicate in the brain.However,since evidence of SARS-CoV-2 occurrence in the brain is not conclusive,most researchers are biased toward thinking of neurological symptoms during the “long COVID” as a consequence of the infection-triggered inflammatory response.Under this view,it is not surprising that treatment with anti-inflammatory drugs in hospitalized COVID patients prevented “long COVID” emergence.Thus,the question is whether SARSCoV-2 induces a primary or secondary encephalopathy linked to several major COVID-linked events: inflammation,hypoxia by respiratory failure,hypoperfusion,assisted ventilation,autoimmunity,and BBB disruption(Ali Awan et al.,2021;de Erausquin et al.,2021).Altered BBB could allow the infiltration of immune cells,which may contribute to cognitive decline(Ciaccio et al.,2021).Activation of microglia,variable degrees of astrogliosis,and infiltration with CD8-positive T cells are the main findings reported in a comprehensive neuropathological study of patients who died from COVID-19(Matschke et al.,2020).As stated by Fleischer et al.(2021),among COVID patients with cerebral ischemia,50% had BBB disruption and increased interleukin levels in CSF,but all CSF tested negative for SARS-CoV-2 RNA.Antibodies against neuronal and glial epitopes were detected in 35% of the patients tested.The authors interpreted these results as neurological compromise driven by inflammation due to BBB disruption and cytokine release,rather than direct viral invasion.The increased risk of cerebrovascular accident is a manifestation of SARS-CoV-2-provoked cytokine storm and coagulation abnormalities (Wu et al.,2020).In particular,hypoxia/ischemia contributes to the degradation of BBB through the activation of matrix metalloproteinases,which break down the basal lamina and ti ght junctions of the endothelium (Yang and Rosenberg,2011).In COVID-19 cases,dysfunction of endothelial cells added to inflammation may lead to abnormalities in coagulation and thrombo-inflammatory processes,promoting vasculopathy(Barbosa et al.,2021).Indeed,endothelial cells express ACE2 and high levels of neuropilin-1,which makes them susceptible to infection by SARS-CoV-2(Wenzel et al.,2021).Besides systemic effects,SARS-CoV-2 infection can induce microvascular alterations,like the formation of string vessels likely derived from endothelial cell death after SARS-CoV-2-induced survival factor degradation (Wenzel et al.,2021).These facts warn us about a possible association of COVID-19 with vascular dementia,which,in turn,shares pathognomonic signs with AD.
Evidence of Alzheimer’s Disease-Like Neuropathology after COVID-19
There is literature indicating that human CoVs can remain dormant in neurons (Arbour et al.,1999) and that COVID-19 associates with generalized gene expression regulation in the brain,which may contribute to its longstanding effects (Yang et al.,2021).Extensive inflammation and degeneration have been shown in COVID-19 brains,including those of people without neurological symptoms,and an overlap was found between marker genes of AD and genes that are upregulated in COVID-19 infection (Yang et al.,2021).Inflammatory biomarkers including interleukin-6,interleukin-1,TNF,complement proteins,and galectin-3 have been proposed as common prognostic biomarkers between SARS-CoV-2 infection and AD,as reviewed in (Ciaccio et al.,2021) and (Rahman et al.,2021).The activation of the NLRP3 inflammasome,triggered during SARS-CoV-2 infection,could lead to downstream tau aggregation and neurodegeneration (Reiken et al.,2022).
Besides viral neurotropism and inflammation resulting from a cytokine storm,infection with SARS-CoV-2 may also initi ate protein aggregation via heparinbinding sites and/or spike-derived peptides,leading to amyloid fibrils just like those in AD (Tavassoly et al.,2020).Elevated levels of Aβ,neurofilament light chain,neurogranin,glial fibrillary acidic protein,and tau were reported in CSF from patients 1–3 months after recovery from COVID-19 (Ciaccio et al.,2021;Sun et al.,2021),showing correlation with neurological symptoms.These markers were also increased in the extracellular vesicles of neural origin of individuals recovering from COVID-19 (with or without self-perceived neurological symptoms) and coincide with extracellular vesicles of neural origin content found in AD (Sun et al.,2021).
Several authors have reported shared gene signatures between AD and COVID-19.Reiken et al.(2022) showed that SARS-CoV-2 infection activates inflammatory signaling and oxidative stress pathways and increases AMPK and GSK3β phosphorylation,resulting in hyperphosphorylation of tau,but normal APP processing in COVID-19 patients’ cortex and cerebellum.There was reduced calbindin expression rendering both tissues vulnerable to Ca2+-mediated pathology.Interestingly,brains from both young and aged patients demonstrated increased tau phosphorylation in the cerebellum,which is not typical of AD (Reiken et al.,2022).Remarkably,Ramani et al.(2020)showed that SARS-CoV-2 enters 3D human brain organoids within 2 days of exposure,mainly targeting neurons,which displayed tau mislocalization,hyperphosphorylation,and apparent cell death.However,and in agreement with the suggestion of Gagliardi et al.(2021) mentioned above,SARS-CoV-2 does not appear to efficiently replicate within the brain organoids.
The Triggering Receptor Expressed on Myeloid cells 2 gene (TREM2) is expressed in microglia and stimulates phagocytosis while suppressing cytokine production and inflammation (Rohn,2013).Gene variants of TREM2 have been linked to AD risk.However,the knockout of TREM2 inhibits neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy (Leyns et al.,2017),suggesting the dual roles of TREM2.Interestingly,Wu et al.(2021) found that TREM2 was induced in T cells in the blood and lungs of patients with COVID-19.TREM2 binds to SARS-CoV-2 membrane protein through its immunoglobulin domain,promoting the T helper cell response.These results also support the association between infecti ous processes and AD risk genes.
COVID-19 and Amyloid-β
The exploratory prospective study of Ziff et al.(2022) found that patients with neurological syndromes within 40 days of symptomatic COVID infection had significantly reduced CSF soluble amyloid precursor protein (sAPP)-ɑ and sAPPβ,as well as Aβ40,Aβ42,and Aβ42/Aβ40ratio compared to controls,indicative of AD risk.Patients with COVID-19 neurological syndromes showed significantly increased neurofilament light chain and proinflammatory cytokines and this negatively correlated with sAPPɑ and sAPPβ CSF levels.Conversely,glial fibrillary acidic protein was significantly reduced in CSF from COVID-19 patients with neurological symptoms,and this positively correlated with sAPPɑ and sAPPβ CSF levels.All CSF samples were SARS-CoV-2 PCR-negative.Lower sAPPα and glial fibrillary acidic protein might indicate astrocyte dysfunction and impaired neuroprotection since astroglial sAPPα is protective against neurotoxicity of Aβ (Durand et al.,2017).
Inflammatory cytokines induce the expression of the interferon-induced transmembrane protein 3 (IFITM3) in neurons and astrocytes,which upregulates γ-secretase activity,thereby increasing the production of Aβ(Hur et al.,2020).But IFITM3 is also a viral particle sequestration protein and SARS-CoV-2 significantly upregulatesIFITM3(Sardar et al.,2020).Comparative transcriptomic studies showed that FYN kinase and IFITM3 biological networks were significantly enriched in several COVID-19 datasets containing SARS-CoV-2 upregulated genes (Vavougios et al.,2021a),priming APP for gamma processing and Aβ generation.After having found pathways and gene signatures shared between AD and COVID infection,Vavougios et al.(2021b) outlined that “IFITM3,exosomal hyperphosphorylated tau,and Aβ-nucleic acid complex may constitute a feed-forward signal expanding from a primary site of neuroinfection via afferent projections;the signal itself would propagate AD pathology even in the absence of infection”.Indeed,Camacho et al.(2021) performed a meta-analysis of the genes regulated in response to SARS-CoV-2 S protein binding to brain microvascular endothelial cells,which helped predict a decrease in APP expression in the early stages of COVID infection and an increase in APP expression in later stages of infection involving inflammatory damage.
On the other hand,Hsu et al.(2021) demonstrated that Aβ42,but not Aβ40,binds to various viral proteins with a preferentially high affinity for the S1 subunit of SARS-CoV-2 spike protein and the viral receptor ACE2,and strengthens the binding of SARS-CoV-2 S1 protein to ACE2,thereby enhancing viral entry.This result clearly objects the anti microbial property of Aβ.Figure 2summarizes all these recent findings.
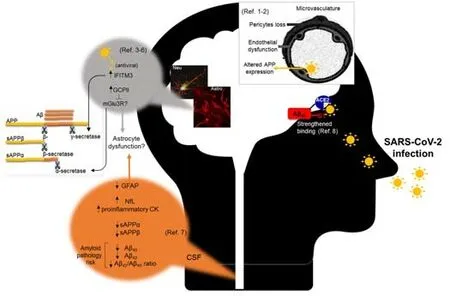
Figure 2|Different pathways and cell types reported involved in the association between SARS-CoV-2 and amyloid pathology.
Proposed Targets for Neurorepair Might Be Altered by SARS-CoV-2
COVID-19 can cause autoimmune reactions and glutamate excitotoxicity(Dolatshahi et al.,2021).Systemic inflammation leads to decreased production of monoamines and trophic factors and activation of microglia,resulting in increased glutamate and N-methyl-D-aspartate levels,leading to excitotoxicity (Boldrini et al.,2021).Dysfunctional astrocytes may add to increased excitotoxicity by impaired glutamate clearance activity.Wang et al.(2021b) showed that pericytes,when integrated into a cortical organoid,are susceptible to infection with SARS-CoV-2 and mediate virus spreading to astrocytes,which exhibit an inflammatory type I response and cell death.In line with a limited capacity of astrocytes to clear excitotoxic glutamate,it has been suggested by Reiken et al.(2022) that subtype 3 metabotropic glutamate receptor (mGlu3R) may be underactivated in astrocytes from COVID brains.The authors reported elevated expression of glutamate carboxypepti dase 2 in COVID-19 brains,which may contribute to increased PKA signaling by inhibiting mGlu3R.Since mGlu3R has been broadly reported to induce astroglial glutamate transporters and reduce excitotoxicity,a failure in mGlu3R activity could contribute to increased excitotoxicity in COVID brains.
Although highly homologous in their sequences,mGlu3 and mGlu2 receptors exhibit relevant functional differences,at least regarding AD involvement.While mGlu3R is neuroprotective against Aβ,mGlu2R was shown to potenti ate Aβ neurotoxicityin vitro(Caraci et al.,2011).Interestingly,it has been recently demonstrated that mGlu2R is an internalization factor for SARS-CoV-2 since it directly interacts with SARS-CoV-2 spike protein,and knockdown of mGlu2R decreases internalization of SARS-CoV-2 but not cell binding.Further,knocking out mGlu2R in mice abolishes SARS-CoV-2 infection in the nasal turbinates and significantly reduces viral infection in the lungs(Wang et al.,2021a).
Conclusions
Evidence for pathogens leading to AD adds to the presumption of long-term neuropsychiatric sequelae of COVID-19.Carefully tracking the neurological impact of COVID-19 emerges as a major scientific challenge.Reported results regarding the cognitive decline in a significant portion of COVID-19 patients,Aβ and tau pathways altered by SARS-CoV-2,neurotoxic glial profile in response to the virus,and brain changes in discrete regions related to learning/memory,made us aware of a possible eti ological link between this viral infection and AD.Although undesirable,the COVID pandemic will indeed allow scienti sts to explore the infecti ous hypothesis prospectively in a wide population.We will also be able to test preventive interventional strategies aimed at promoting synaptic plasticity that may overcome COVID-induced brain damage within an appropriate time window to delay late-onset AD.Up to May 2022,a search for “SARS CoV 2 infection AND cognitive” terms on the clinicaltrials.gov site yielded 90 clinical trials,whereas the terms “SARS CoV 2 infection cognitive long term” yielded 11 results,with none planning for long follow-up periods.This invites us to subscribe to the concern of Itzhaki et al.(2016) about neglected clinical prospective studies on pathogen-driven neurodegeneration.
Author contributions:Conceptualization: DD;data curation: EO,AS,DD;writing– original draft : EO,AS,DD;writing– review and editing: LC,CC,ML;funding acquisition: DD.All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Open access statement:This is an open access journal,andarticles are distributed under the terms of the Creative Commons Attribution Non Commercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Kevin N Hascup,Southern Illinois University School of Medicine Neurology,USA.
Additional file:Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Bystanders or not? Microglia and lymphocytes in aging and stroke
- Serine and arginine rich splicing factor 1: a potenti al target for neuroprotection and other diseases
- Can glial cells save neurons in epilepsy?
- Lights for epilepsy: can photobiomodulation reduce seizures and offer neuroprotection?
- The landscape of cognitive impairment in superoxide dismutase 1-amyotrophic lateral sclerosis
- CMT1A current gene therapy approaches and promising biomarkers