
Converging links between adult-onset neurodegenerative Alzheimer’s disease and early life neurodegenerative neuronal ceroid lipofuscinosis?
2023-02-24 05:24MarcelKleinGuidoHermey
中国神经再生研究(英文版) 2023年7期
Marcel Klein,Guido Hermey
Abstract Evidence from genetics and from analyzing cellular and animal models have converged to suggest links between neurodegenerative disorders of early and late life.Here,we summarize emerging links between the most common late life neurodegenerative disease,Alzheimer’s disease,and the most common early life neurodegenerative diseases,neuronal ceroid lipofuscinoses.Genetic studies reported an overlap of clinically diagnosed Alzheimer’s disease and mutations in genes known to cause neuronal ceroid lipofuscinoses.Accumulating data strongly suggest dysfunction of intracellular trafficking mechanisms and the autophagy-endolysosome system in both types of neurodegenerative disorders.This suggests shared cytopathological processes underlying these different types of neurodegenerative diseases.A better understanding of the common mechanisms underlying the different diseases is important as this might lead to the identification of novel targets for therapeutic concepts,the transfer of therapeutic strategies from one disease to the other and therapeutic approaches tailored to patients with specific mutations.Here,we review dysfunctions of the endolysosomal autophagy pathway in Alzheimer’s disease and neuronal ceroid lipofuscinoses and summarize emerging etiologic and genetic overlaps.
Key Words:Alzheimer’s disease;autophagy;Batten disease;CLN3 disease;dementia;endosome;lysosome;neurodegeneration;neuronal ceroid lipofuscinosis;presenilin
Introduction
Neurodegenerative diseases of early and late life demonstrate distinct clinical signatures and have traditionally been viewed independently.Early life neurodegenerative disorders are usually monogenetic and rare diseases.Whereas adult-onset neurodegenerative disorders are frequently genetically complex,with a high incidence rate among older adults,in several cases sporadic,and additionally driven by live style,environmental factors and therefore epigenetic modifications.In addition to the differential time of diagnosis and varying pathological hallmarks,neurodegenerative diseases can be categorized as disorders of specifically affected cellular pathways or organelles.Several monogenetic disorders have been classified by striking pathological abnormalities observed by biochemistry,cell biology or microscopy and include disorders of carbohydrate or amino acid metabolism,peroxisomal,mitochondrial or lysosomal disorders and the recently grouped autophagy disorders (Ballabio and Gieselmann,2009;Ebrahimi-Fakhari et al.,2016;Sheng,2017;Darios and Stevanin,2020;Deb et al.,2021;Myerowitz et al.,2021).Several adult-onset neurodegenerative disorders show specific progressive abnormalities in varying organelles and can therefore also be classified accordingly.
Links between Parkinson’s disease and Gaucher disease
Evidence from genetics and cellular and animal model investigations converged to suggest links between specific monogenetic early life and late life neurodegenerative diseases.An example of a rare disease linked to a common neurodegenerative disease is Gaucher disease (GD),a lysosomal storage disease that manifests early in life,which has been linked to Parkinson’s disease (PD),one of the most common adult-onset neurodegenerative disorders.Epidemiological investigations disclosed hereditable forms causing familial autosomal dominant PD by mutations in the α-synuclein geneSNCAorLRRK2(de Lau and Breteler,2006;Minakaki et al.,2020) and early onset autosomal recessive PD by mutations inPARKIN,PINK1orDJ-1(de Lau and Breteler,2006;Minakaki et al.,2020).However,the majority of PD cases are regarded as sporadic and arise from an interplay of genetic and environmental factors.Today,known as the most common genetic risk factor for PD are heterozygous mutations in the glucocerebrosidase (GCase) geneGBA1(Sidransky et al.,2009).GCase is a lysosomal hydrolase that catalyzes the breakdown of glucosylceramide to glucose and ceramide.HeterozygousGBA1variants are thought to influence PD pathogenesis through multiple mechanisms.These include accumulation of misfolded GCase in the endoplasmic reticulum (ER),impairing ERquality control,and its accumulation on the surface of lysosomes,inhibiting chaperone-mediated autophagy (CMA),resulting in impaired protein degradation and accumulation of GCase substrates (Ron and Horowitz,2005;Mazzulli et al.,2011;Kuo et al.,2022a,b).Prior to its discovery as a PD risk locus,homozygous (or bi-allelic) missense mutations ofGBA1were known to cause the autosomal recessive disorder GD (Roh et al.,2022).It results from GCase deficiency and the accumulation of its substrates within lysosomes leading to lysosomal dysfunction and impairments in the autophagy pathway(Roh et al.,2022).Due to the defective acti vity of a lysosomal protein and the accumulation of lysosomal storage material,GD is classified as a lysosomal storage disease and regarded as the most common form (Roh et al.,2022).Interestingly,there is also recent evidence for the involvement of additional lysosomal storage disease genes in PD risk (Robak et al.,2017;Huebecker et al.,2019).Moreover,patients affected by GD have a higher risk of PD (Goker-Alpan et al.,2004).Collectively,these findings classify PD on the cellular level also as a lysosomal disease.Different therapeutic strategies have been developed to treat GD employing enzyme replacement therapy and substrate reduction therapy involving small molecules (Do et al.,2019;Ivanova et al.,2021).Some of these strategies might actually as well be beneficial for PD patients.Congruously,a number of strategies targeting the GCase ceramide metabolism to improve autophagy-lysosome dynamics have been suggested to be therapeutic for both GD and PD (Bonam et al.,2019;Ysselstein et al.,2019).Together,findings from the last decades demonstrate how insights from a rare monogenic early life neurodegenerative disorder,such as GD,can direct research into the pathogenesis and therapy of a seemingly unrelated common and complex neurodegenerative disorder of late life such as PD.This raises the intriguing question whether there are also links between other late life and specific monogenetic early life neurodegenerative diseases.
Links between Alzheimer’s disease and neuronal ceroid lipofuscinosis?
We recently reported biochemical and cellular evidence for converging roles of presenilin enhancer (PSENEN or PEN2) and ceroid-lipofuscinosis neuronal 3 (CLN3) in the autophagy-lysosome system (Klein et al.,2022).PEN2 is a subunit of the γ-secretase complex whose dysfunction relates to the adult-onset neurodegenerative disorder Alzheimer’s disease (AD).Mutations in CLN3 are causative for CLN3 disease which belongs to the neuronal ceroid lipofuscinoses (NCLs),also named Batten disease,a group of neurodegenerative disorders of early life.On the cellular and molecular level,a growing number of independent studies strongly suggest dysfunction of intracellular trafficking and sorting mechanisms as well as of the autophagylysosome system in both types of neurodegenerative disorders (Festa et al.,2021;Kim et al.,2022).A better understanding of the shared mechanisms underlying the different diseases is important as this might lead to the identification of novel targets for therapeutic development,the transfer of therapeutic strategies from one disease to the other and therapeutic strategies tailored to patients with specific mutations.
In this review,we will highlight emerging links between the adult-onset neurodegenerative disorder AD and the early-onset neurodegenerative disorders NCL suggesting shared cytopathological processes underlying these different neurodegenerative diseases.We will first briefly introduce intracellular endosomal sorting and trafficking and the lysosome-autophagy system.We will then review dysfunction of these pathways in AD and NCL,and summarize eti ologic and genetic overlaps.
Retrieval Strategy
To examine the published links between NCL and AD,we conducted a search of PubMed and Google Scholar between May and August 2022 without the restriction of publication dates for neurodegeneration,Alzheimer’s disease,neuronal ceroid lipofuscinosis,Batten disease,CLN,intracellular sorting and trafficking,lysosome and autophagy.
Autophagy-Endolysosome System
Proteins are delivered post-translationally to specific subcellular compartments to exert their proper function.Protein sorting in the exo-and endocytic pathway occurs at three major sites,the Golgicomplex,endosomes and the plasma membrane (Figure 1).Active transport of proteins is mediated by tubular or vesicular intermediates that bud from one compartment and fuse with the next.Adaptor proteins recognize sorting signals,short amino acid sequences in the cytosolic domains of transmembrane proteins,and convey selective transport (Bonifacino,2014).Specific transmembrane proteins bind ligands through their luminal/extracellular domain and bind through their cytoplasmic domain adaptor proteins,which recruit additional components such as clathrin scaffolds and accessory proteins and direct receptor-ligand complexes to specific vesicle carriers (Braulke and Bonifacino,2009).Secretory proteins synthesized in the ER are transported to the Golgicomplex,sorted to secretory vesicles at thetrans-Golgi network and targeted to the plasma membrane through regulated or constitutive pathways.At the plasma membrane transmembrane proteins can be internalized,enter early endosomes and segregate into separate trafficking itineraries.Transmembrane proteins can be recycled back to the plasma membrane through fast or slow recycling compartments.An alternative route directs internalized cargo from early endosomes to late endosomes,so-called multivesicular bodies,and retrogradely to thetrans-Golgi network or further to lysosomes for degradation (Meraş et al.,2022;Figure 1).The majority of soluble lysosomal enzymes are modified with mannose 6-phosphate,allowing their recognition by mannose 6-phosphate receptors in the Golgi apparatus and their targeting to lysosomes.Other soluble lysosomal proteins are transported in a mannose 6-phosphate independent manner by alternative receptors,such as the lysosomal integral membrane protein 2(LIMP2) or Sorti lin (Braulke and Bonifacino,2009).Lysosomes are degradative compartments that break down intracellular and exogenous substrates into their constituent building blocks.Pathogens,defective organelles or proteins and protein aggregates can be degraded by autophagy.Different types of autophagy are known,including macroautophagy,CMA and microautophagy.All route material to lysosomes for degradation (Fleming et al.,2022).Microautophagy mediates lysosomal degradation of cellular components through membrane invaginations in compartments of the endolysosomal system.In CMA,substrate proteins are targeted by the recognition of a KFERQ-like amino acid moti f and translocated across the lysosomal membrane through a multimeric complex including the lysosome-associated membrane protein type 2A (LIMP-2A).Macroautophagy is an inducible catabolic process in which double membrane-bounded autophagosomes are formed around substrates and eventually fuse with lysosomes to autolysosomes for substrate degradation.The microtubule-associated protein 1 light chain 3 (LC3) plays key roles in macroautophagy and is frequently used as a marker protein for autophagosomes (Ballabio and Bonifacino,2020).Lysosomes function also as metabolic signaling hubs.A key regulator of lysosomal function,nutrient sensing and regulation of gene expression is mammalian target of rapamycin complex 1 (mTORC1) that integrates nutrient availability and growth factor signaling.Downstream signaling of the multi protein complex mTORC1 includes induction of autophagy and regulation of translation and transcription factors.Activation and inhibition of these different pathways are ti ghtly regulated by phosphorylation of the different signal transducers.The transcription factor EB coordinates the expression of lysosomal and autophagy proteins in response to pathways sensing lysosomal stress and nutritional conditions (Ballabio and Bonifacino,2020).Transcription factor EB localizes to the cytosol.Upon activation,it translocates to the nucleus and initi ates gene expression.
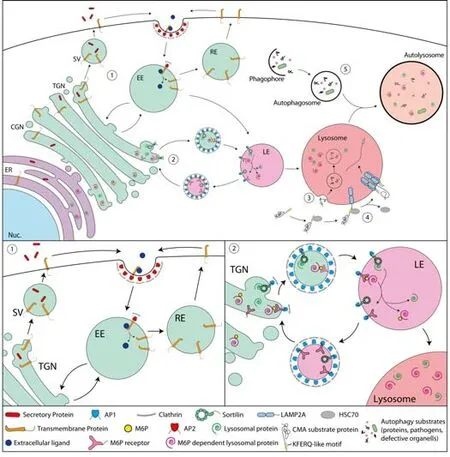
Figure 1|Endosomal sorting and the lysosome-autophagosome system.
Alzheimer’s Disease
AD is the most prevalent progressive adult-onset neurodegenerative disorder characterized by cognitive and memory dysfunction.Hallmark histopathologies are intraneuronal neurofibrillary tangles,comprising the microtubule binding protein Tau,and extracellular amyloid plaques,composed of proteolytically cleaved amyloid β peptides (Aβ),which are fragments of the amyloid precursor protein (APP) (DeTure and Dickson,2019).Genetic evidence strongly suggests that alterations in proteolytic APP processing have a significant impact on AD pathology.Dominantly inherited familial AD that manifests in the early twenties,with an average age of disease onset in the mid-forti es,accounts for~1% of AD cases (DeTure and Dickson,2019).There are also other early onset forms of AD characterized by developing symptoms after 50 years and dementi a in the mid-60s.Late onset AD is the most common form,accounting for~95% of AD cases,regarded as sporadic and multi factorial with a high level of heritability.
The autophagy-endolysosome system in AD
Genomic studies identified genes linked to the development of the early onset familial forms and a larger number of genetic factors associated with increased risk for developing late onset AD.The encoded proteins have been functionally assigned to different biological pathways.Among the identified genetic variations,those found in genes coding for proteins related to the autophagy-endolysosome network attracted much attention,because of known prominent neuropathological features of the autophagy-endolysosome system in AD (Cataldo et al.,2000;Whyte et al.,2017).In addition,several studies strongly suggest that the genes in which mutations are causal for rare autosomal forms of AD encode proteins playing a role in the endosomallysosomal system (Wolfe et al.,2013;Nixon,2017;Fleming et al.,2022;Szabo et al.,2022).
The prominent neuropathological features of the autophagy-endolysosome system in AD comprise an increase in size and volume of early endosomes,lysosomes and autophagic vacuoles,and an accumulation of lysosomal hydrolases,such as cathepsin D (CTSD) (Cataldo et al.,1991;Nixon,2017;Whyte et al.,2017;Lee et al.,2022b).These findings suggest the deregulation of the autophagy-endolysosome network in AD.Conversely,a gradual loss of CMA with age may be a risk factor for AD.CMA is inhibited in experimental models of tauopathies,in AD patient brains at an early disease stage and loss of CMA in a mouse model of AD accelerated disease progression (Bourdenx et al.,2021).Moreover,CMA deficiency increases the similarity between the proteomes of brains from an AD mouse model and AD patients,thus mimicking part of the disease.Furthermore,upregulation of CMA in two different mouse models of AD ameliorates the disease phenotype (Bourdenx et al.,2021).
Genes associated with familial AD
Mutations inAPP,PSEN1,andPSEN2are causal for rare autosomal familial forms of AD and mutations inSorL1underlie additional rare early onset variants.APP,a type-I transmembrane protein,is sequenti ally processed by different enzymes in alternative pathways (Eggert et al.,2018;Checler et al.,2021).In one pathway,cleavage by β-secretase in the luminal/extracellular part produces the membrane embedded β-C-terminal fragment,also named C99.γ-Secretase cleaves this fragment and releases Aβ and the intracellular domain of APP.The proteolytic processing by β-secretase and subsequently by γ-secretase,regarded as the amyloidogenic pathway,occurs during APP’s itinerary through the endolysosomal system.It has gained the most attention because it results in the plaque forming Aβ and mutations in APP and components of the γ-secretase have been genetically linked to familial forms of AD.γ-Secretase is composed of four transmembrane proteins PEN2,NCSTN,APH1 and presenilin 1 (PSEN1) or 2 (PSEN2),which harbor the catalytic site.Several mutations inPSEN1andPSEN2increase the production of Aβ.Cells co-express differing γ-secretase complexes including PSEN1 or PSEN2.PSEN1-containing complexes are broadly distributed in the cell whereas PSEN2 is mainly targeted to late endosomes and lysosomes(Meckler and Checler,2016;Sannerud et al.,2016).In agreement,neuronal acidic compartments present γ-secretase activity (Maesako et al.,2022).Introduction of disease-causing mutations in or genetic ablation ofPSEN1orPSEN2or depleting formation of γ-secretase byPEN2ablation results in altered lysosomal acti vity and accumulation of autophagosomes (Lee et al.,2010;Neely et al.,2011;Whyte et al.,2017;Fedeli et al.,2019;Klein et al.,2022).Initi al studies proposed γ-secretase independent functions of PSENs in the autophagy-lysosome system because pharmacological inhibition of γ-secretase acti vity did not induce similar phenotypes as the genetic ablation,such as enlarged lysosomes and accumulation of the autophagosome marker LC3-II (Wilson et al.,2004;Neely et al.,2011).In contrast,other studies demonstrated the increased size of early endosomes,disrupted lysosomal proteolysis and autophagic impairment after pharmacological inhibition of γ-secretase (Jiang et al.,2010;Lauritzen et al.,2016;Hung and Livesey,2018;Kwart et al.,2019).The congruent phenotype observed by genetic ablation,and pharmacological inhibition of PSENs suggests that the loss of γ-secretase activity is actually causing at least some of the observed alterations in the autophagy-endolysosome system.Notably,the introduction of mutations inAPPandPSEN1causal for autosomal dominant early-onset monogenic AD led to converging defects in the autophagy-endolysosome system in human induced pluripotent stem cell (iPSC)-derived neurons (Hung and Livesey,2018;Kwart et al.,2019).Checler and colleagues hypothesized that the PSEN1 phenotype relates to the accumulation of the C99 APP fragment,as β-secretase,but not γ-secretase,inhibition rescues the phenotype (Checler et al.,2021).Interestingly,C99 induces autophagy-lysosome impairments independently of Aβ (Lauritzen et al.,2016) and contains a KFERQ moti f that could be uti lized for its degradation through CMA (Fleming et al.,2022).
The sortilin-related receptor 1 (Sorl1) gene encodes SorLA,an established sorting receptor for APP,promotes retrograde sorting of APP from early endosomes to thetrans-Golgi network and directs APP to a nonamyloidogenic pathway (Andersen et al.,2006).SorL1haploinsufficiency causes enlarged endosomes (Andersen et al.,2022) andSorL1ablation results in endosomal trafficking impairments and defects in the autophagyendolysosome network (Knupp et al.,2020;Hung et al.,2021;Mishra et al.,2022).In conclusion,the genes linked to rare early onset forms of AD,APP,PSEN1,PSEN2andSorL1,encode proteins with proposed roles in the autophagy-endolysosome system.
Genes associated with sporadic AD
A large number of additional genes have been associated with AD risk and several of these relate to the autophagy-endolysosome system,too (Szabo et al.,2022).Among the encoded proteins are the SorLA related sorting receptors Sorti lin and the sorti lin related VPS10 domain containing receptor 1 (SorCS1) that also interact with APP,and the cytosolic regulators of endocytosis and trafficking,Bin1 and CALM (Reitz et al.,2011;Gustafsen et al.,2013;Hermey et al.,2015;Bellenguez et al.,2022).
Differentiated neurons with their long axons and complex dendritic trees are highly vulnerable to changes in the endolysosomal system,because they depend on functional long-range intracellular endosomal transport for proper function.In addition,neuronal cells are post mitotic and cannot dilute accumulated proteins by cell division.Thus,the defects in the autophagy-endolysosome system underlying AD may have a modest impact on autophagy-endolysosome function,but result in a long-term effect over lifetime probably in incorrect protein targeting,reduced degradation and accumulation of protein aggregates and finally cellular degeneration.
Together,AD occurs as sporadic forms with complex genetics or as monogenetic forms which have varying time points of manifestation.Different cellular mechanisms causing AD may exist in parallel,but accumulating evidence suggest that the autophagy-endolysosome pathway is critical to the pathology of AD.
Neuronal Ceroid Lipofuscinosis
NCLs,commonly named Batten disease,are a group of rare lysosomal storage disorders mainly affecting children and are considered the most common neurodegenerative disease in early life (Kohlschütter et al.,2019;Butz et al.,2020).NCLs are characterized by gradual neurodegeneration,including seizures and progressive loss of vision,motor function and cognition,and eventually premature death.The pathological hallmark is intracellular accumulation of autofluorescent lysosomal storage material,socalled lipofuscin (Mole et al.,2005;Radke et al.,2015).These deposits are found in neurons,but are also abundant in non-neuronal cells outside the nervous system.Depending on the subtype,the storage material is either predominantly composed of subunit C of the mitochondrial ATP synthase(SCMAS) or the sphingolipid activator proteins A and D (Mole et al.,2005;Radke et al.,2015;Butz et al.,2020).The storage material can comprise to a different degree additional components such as lysosomal proteins like palmitoyl protein thioesterase 1 (PPT1),tripeptidyl peptidase 1 (TPP1) and CTSD (Anderson et al.,2013),and strikingly,accumulation of Aβ has been reported in some cases (Wisniewski et al.,1990a;Wisniewski et al.,1990b;Herva et al.,2000).The age of disease manifestation differs between defined subtypes caused by mutations in different genes (Camp and Hofmann,1993;Vesa et al.,1995;Wang et al.,1998;Lin et al.,2001;Steinfeld et al.,2006;Kohlschütter et al.,2019;Butz et al.,2020;Lee et al.,2022a;Table 1).The NCLs comprise 13 monogenetic diseases,which are all autosomal recessive and caused by homozygous or compound heterozygous mutations with the exception of the autosomal dominant CLN4 that is caused by heterozygous mutation in theDNAJC5gene (Kohlschütter et al.,2019).NCLs are thought to have parti ally shared,but also unique pathomechanisms involving alterations in the autophagy-endolysosome system (Huber,2020;Nelvagal et al.,2020).In agreement,the majority of the disease-causing genes encode proteins with a predominant lysosomal localization,but some are located in other cellular compartments (Table 1).
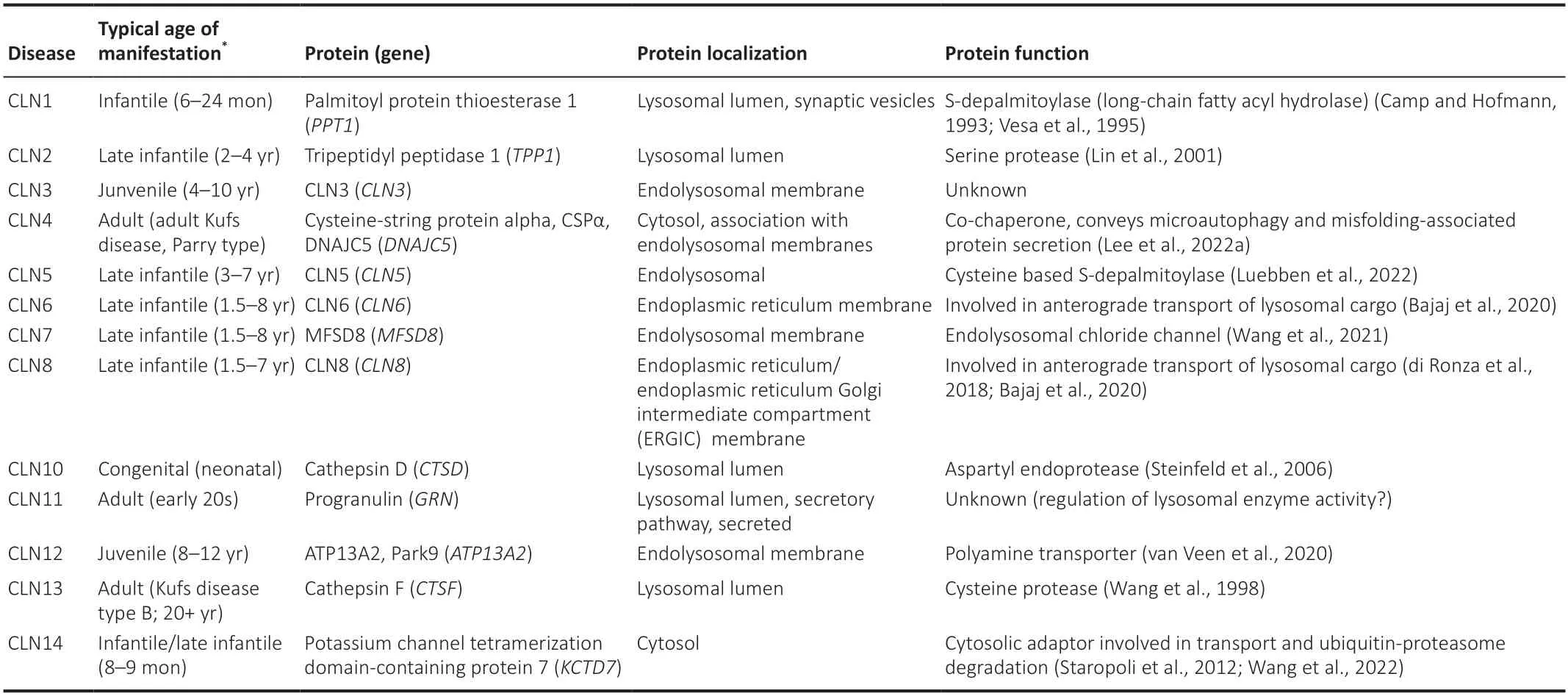
Table 1|Neuronal ceroid lipofuscinoses
Soluble CLN proteins
The CLN proteins can be classified into soluble and transmembrane proteins.The group of soluble proteins comprises lysosomal and cytosolic proteins.The lysosomal enzymes PPT1 (CLN1),TPP1 (CLN2),CTSD (CLN10) and cathepsin F (CTSF) (CLN13) are targeted directly from the Golgi to lysosomes where these hydrolases are activated (Butz et al.,2020).Whereas progranulin(granulin precursor,PGRN,encoded by the geneGRN) (CLN11) is either directly targeted from the Golgi to lysosomes or alternatively secreted to the extracellular space.Here,PGRN may be proteolytically processed to smaller peptides,so-called granulins,that possess function independent and sometimes in contrast to the granulin precursor,e.g.as a growth factor(Paushter et al.,2018).PGRN is translocated to lysosomes through direct interaction with the endocytic sorting receptor Sortilin (Hu et al.,2010) or through interaction with prosaposin and its receptors,mannose 6-phosphate receptor or low-density lipoprotein receptor-related protein 1 (LRP1),that mediate reuptake and intracellular targeting of the interactors to lysosomes(Zhou et al.,2015).Accumulating evidence suggests that lysosomal PGRN directly or indirectly regulates the acti vity of lysosomal enzymes such as CTSD or GCase (Paushter et al.,2018).Several soluble lysosomal CLN proteins,such as PPT1,TPP1,CTSD and CTSF,have been also detected extracellularly where these may serve additional functions (Huber,2021).
Two soluble CLN proteins are cytosolic,DNAJC5 (CLN4) and KCTD7 (CLN14).DNAJC5 associates with endosomal and lysosomal membranes or perinuclear compartments (Benitez et al.,2011;Nosková et al.,2011;Lee et al.,2022a).Endolysosomal-associated DNAJC5 promotes ESCRT-dependent microautophagy and non-lysosomal DNAJC5 conveys misfolding-associated protein secretion (Lee et al.,2022a).KCTD7 ablation reduces lysosomal enzyme trafficking and causes lysosomal and autophagic defects (Wang et al.,2022).KCTD7 belongs to the BTB domain-containing adaptor subfamily(KCTD for potassium channel tetramerization domain).Several BTB domaincontaining adaptors of the KCTD family act as substrate binding receptors for Cullin-3,a scaffolding protein component of E3 ubiquitin-ligase complexes that selectively tag proteins for degradation by the proteasome.Similarly,KCTD7 interacts with Cullin-3 and CLN14 patient-derived missense mutations in the BTB domain of KCTD7 disrupt the interaction with Cullin-3 (Staropoli et al.,2012).Moreover,KCTD7 serves as a Cullin-3 adaptor to recruit CLN5 to the ubiquiti n-proteasome pathway (Wang et al.,2022).
Transmembrane CLN proteins
The remaining six CLN proteins are transmembrane proteins.CLN5 is a type-II transmembrane protein with a cytoplasmic N-terminus,one transmembrane domain and a large luminal C-terminal moiety.CLN5 is proteolytically processed (De Silva et al.,2015) and converted to a soluble form by signal pepti dase and homologous signal pepti dase-like proteases (Jules et al.,2017).One report suggested that CLN5 functions as a glycoside hydrolase,but endogenous substrates have not been identified (Huber and Mathavarajah,2018).However,a more recent study identified CLN5 as a lysosomal cysteinebased S-depalmitoylase with the catalytic activity in the processed luminal moiety (Luebben et al.,2022).The other CLN transmembrane proteins are multi -spanning membrane proteins.These include the ER localized CLN6 and CLN8 that facilitate anterograde transport of lysosomal cargo from the ER to the Golgi (di Ronza et al.,2018;Bajaj et al.,2020).Moreover,CLN3 andMFSD8 (CLN7) are mainly localized to lysosomes (Kyttälä et al.,2004;Storch et al.,2004;Steenhuis et al.,2010;Brandenstein et al.,2016;Oetjen et al.,2016).The function of CLN3 is sti ll elusive.CLN3 has been constantly linked to the regulation of endosomal transport (Uusi-Rauva et al.,2012;Lojewski et al.,2014;Schmidtke et al.,2019;Yasa et al.,2020,2021).Moreover,CLN3 loss of function studies demonstrate a role in lysosomal function,autophagy and phagocytosis (Cao et al.,2006;Chandrachud et al.,2015;Palmieri et al.,2017;Schmidtke et al.,2019;Zhong et al.,2020;Tang et al.,2021;Klein et al.,2022;Scotto Rosato et al.,2022).MFSD8 ablation causes alterations in lysosomal size,motility and exocytosis and impairs autophagy (Brandenstein et al.,2016;Danyukova et al.,2018;von Kleist et al.,2019;Lopez-Fabuel et al.,2022) and a function as an endolysosomal chloride channel has been recently reported(Wang et al.,2021).The transmembrane protein ATP13A2 (CLN12) is a member of the P5 subfamily of ATPases with a late endolysosomal localization(Ramirez et al.,2006).It functions as a polycation transporter that exports polyamines,specifically spermine,from lysosomes into the cytosol (van Veen et al.,2020).
Disease phenotypes and age of manifestation
Together,the CLN genes encode proteins that function in subcellular endosomal trafficking of lysosomal components or act in the lysosomal degradative pathway.Several NCL disease causing mutations in these proteins likely result in loss of function or dominant negative variants,in reduced lysosomal function and are associated with a characteristic disease phenotype with a typical age of manifestation (Table 1).With extending the use of exome sequencing in genetic disease diagnosis,it is more and more recognized that some mutations result in variable disease onset,severity,and progression and even in a clinically distinct phenotype (Kousi et al.,2012a;Butz et al.,2020).Several mutations underlie specific subphenotypes within the NCLs and can lead to a protracted course of the disease.For example,mutations inCLN1usually underlie classical infanti le NCL with disease onset in the second half of the first year of life,but rare mutations inCLN1can cause also late infanti le NCL with first symptoms occurring between 2 and 4 years of age,juvenile NCL with disease manifestation between 5 and 16 years and particularly rare adult forms (Kohlschütter et al.,2019).
Links to other neuronal diseases
Notably,several NCL genes have been linked to other neuronal diseases with an alternative clinical diagnosis (Table 2).Compound heterozygous mutations in theTPP1gene (CLN2) underlie a rare recessive type of spinocerebellar ataxia (Sun et al.,2013;Dy et al.,2015).Non-syndromic retinal degeneration and autophagic vacuolar myopathy have been also linked to mutations inCLN3.Specific homozygous or compound heterozygous mutations inCLN6lead to the development of the adult-onset Kufs type-A disease,a form of NCL without vision loss.These mutations appear to be mutually exclusive with those that underlie CLN6 disease (Arsov et al.,2011;Berkovic et al.,2019).Non-syndromic macular dystrophy can be caused by compound heterozygous mutations inMFSD8(CLN7) (Roosing et al.,2015).The gene has been also identified as a candidate risk factor for frontotemporal dementia (Geier et al.,2019) and amyotrophic lateral sclerosis (Huang et al.,2021).A form of progressive epilepsy with mental retardation,is caused by a mutation in the CLN8 gene (Ranta et al.,1999).CTSD(CLN10) has been genetically linked to PD (Robak et al.,2017).Homozygous or compound heterozygous mutations inATP13A2(CLN12) cause the Kufor-Rakeb syndrome,a rare autosomal recessive form of juvenile-onset atypical PD (PARK9) (Ramirez et al.,2006) and biallelic mutations in ATP13A2 underlie autosomal recessive spastic paraplegia 78 (Estrada-Cuzcano et al.,2017).Homozygous or compound heterozygous mutations inKCTD7(CLN14) cause progressive myoclonic epilepsy-3,without NCL-type lysosomal storage (Van Bogaert et al.,2007;Kousi et al.,2012b;Krabichler et al.,2012).
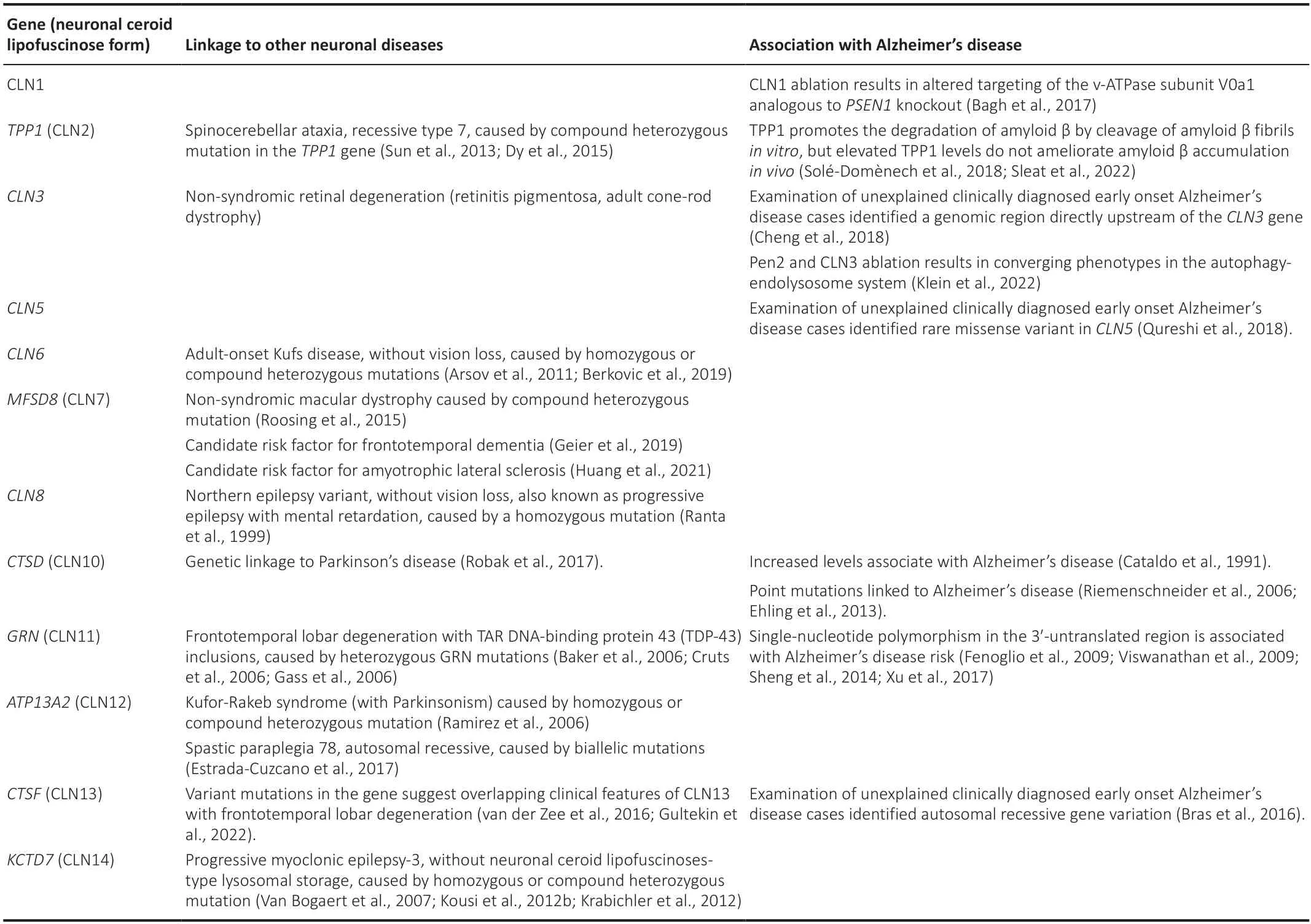
Table 2|Other diseases linked to CLN genes and their association with Alzheimer’s disease
GRN(CLN11) reveals an obvious link to late life neurodegeneration.Heterozygous mutations inGRN,resulting in haploinsufficiency,are the cause of frontotemporal lobar degeneration (FTLD) with TAR DNA-binding protein 43 (TDP-43) inclusions (Baker et al.,2006;Cruts et al.,2006;Gass et al.,2006).FTLD is a clinically heterogenous neurodegenerative disease characterized by progressive atrophy of the frontal and temporal lobes of the brain.Most often,it presents drastic alterations in behavior and personality,including social disinhibition as well as gradual decline in language capabilities.FTLD can be classified by clinical presentation,but also by common histopathological features which comprise inclusion bodies positive for tau protein (FTLD-Tau),ubiquitinated TDP-43 (FTLD-TDP) or fused-in sarcoma protein (FTLD-FUS).GRNmutations are primarily associated with the FTLD-TDP subtype.GRNmutations account for~10% of all FTLD cases and~20–25% of familial FTLD cases.NCL-related phenotypes,specifically elevated levels and accumulation of the NCL-like storage material,were observed in FTLD patients withGRNmutations (Götzl et al.,2014;Valdez et al.,2017;Ward et al.,2017) and some NCL-like features occurred already before dementi a onset (Ward et al.,2017).These observations in autosomal dominant and recessive mutations point at converging pathological mechanisms underlying CLN11 disease and FTLD and dosage effects of PGRN are highly likely to playing a role in disease manifestation.Interestingly,a novel homozygous pathogenic variant in theCTSFgene was identified in a patient diagnosed with FTLD accompanied by parkinsonism (Gultekin et al.,2022) and,independently,heterozygous mutations in theCTSFgene were identified in FTLD patients (van der Zee et al.,2016),suggesting overlapping clinical features of CLN13 disease with FTLD.
Together,the autophagy-endolysosome pathway is critical to the pathology of NCLs.Some of the monogenetic NCLs present varying time points of manifestation.Independent mutations in several NCL genes can cause additional,clinically differenti ally diagnosed neuronal diseases.
Converging Links between Alzheimer’s Disease and Neuronal Ceroid Lipofuscinosis
There is independent supporting evidence for an eti ologic overlap between NCLs and AD.One arises from a study by Dolzhanskaya et al.(2014).A family with progressive dementia,motor deficits in their early 30s and lipofuscin containing phagocytic cells with distinct curvilinear lysosomal inclusion bodies was suspected to have CLN4 disease,but was negative for mutations inDNAJC5.Whole exome sequencing revealed a previously unknown mutation inPSEN1.Thus,the reportedPSEN1mutation is associated with an unusually early onset dementia with a severe fast progressing disease course and lysosomal inclusions,previously believed to be specific for NCL (Dolzhanskaya et al.,2014).
Links from unexplained early onset AD cases
Examinations of unexplained early onset AD cases without clear inheritance patterns or mutations inAPP,PSEN1orPSEN2and with two or more individuals with early onset AD identified a genomic region directly upstream of theCLN3gene using genetic linkage analysis (Cheng et al.,2018) and a missense variant inCLN5by whole-exome sequencing (Qureshi et al.,2018).In addition,GRN(CLN11) has been linked to AD pathophysiology.A singlenucleotide polymorphism in the 3′-untranslated region ofGRNhas been associated with AD risk (Fenoglio et al.,2009;Viswanathan et al.,2009;Sheng et al.,2014;Xu et al.,2017) and single nucleotide polymorphisms in intronic regions ofGRNhave been shown to increase the susceptibility for AD (Viswanathan et al.,2009).Moreover,an autosomal recessiveCTSF(CLN13) gene variation in a clinically diagnosed early-onset AD family has been reported (Bras et al.,2016) and point mutations within theCTSD(CLN10)gene were linked to AD (Riemenschneider et al.,2006;Ehling et al.,2013).The latter findings are consistent with abnormal immunoreacti vity of CTSD in senile plaques of AD patients,which already suggested a molecular link between CTSD and AD (Cataldo et al.,1991).However,the exact role of CTSD dysfunction in disease progression is not well understood,but investigations suggest that AD-associated variants of CTSD are enzymatically active,whereas CLN10-associated variants exhibited impairments of either CTSD maturation and/or enzymatic function (Bunk et al.,2021).
Links from cellular studies
In addition,cellular studies also suggest links between AD and NCL.Bagh and colleagues observed inCLN1ablated cells an altered targeting of the v-ATPase subunit V0a1 resulting in an endolysosomal phenotype analogous to that found inPSEN1knockouts (Lee et al.,2010;Bagh et al.,2017).TPP1 promotes the degradation of Aβ by cleavage of Aβ fibrilsin vitroand this has been proposed as a therapeutic strategy to treat AD (Solé-Domènech et al.,2018).However,a subsequent study by the same research group demonstrated that elevated levels of TPP1 do not ameliorate pathogenesis in a mouse model of AD (Sleat et al.,2022).Early cellular studies suggested that overexpression of CLN3 modulates APP processing (Golabek et al.,2000).Our recent analysis demonstrates thatCLN3ablation does not alter Aβ production or γ-secretase acti vity (Klein et al.,2022).However,we observed an interaction of the γ-secretase subunit Pen2 and CLN3,their co-expression,subcellular co-localization and endosomal co-transport.Using isogenic knockout cells forPen2andCLN3respectively,we observed corresponding cellular alterations in the autophagy-endolysosome system (Klein et al.,2022).These include the reduced acti vity of lysosomal enzymes,an increased number of autophagosomes,increased lysosome-autophagosome fusion,and elevated levels of transcription factor EB.Our study strongly suggests converging roles of Pen2 and CLN3 in the autophagy-lysosome system supporting the idea of common cytopathological processes underlying an early and a late life neurodegenerative disease.Taken together genetic studies suggest an overlap of clinically diagnosed AD and mutations in CLN genes andvice versain one case clinically diagnosed NCL and a mutation in an AD gene.Moreover,cellular studies propose that NCL and AD risk genes functionally converge on the autophagy-endolysosome pathway.This convergence could allow for the buildup of shared therapeutic concepts and tools or the transfer of therapeutic strategies from one disease to the other.Such strategies may comprise pharmacological targeting of lysosome and autophagosome function,e.g lysosomal biogenesis or exocytosis.Accordingly,recent studies focused on the beneficial use of compounds modulating these pathways in NCL disease models (Soldati et al.,2021;Scotto Rosato et al.,2022).
Conclusion
Convergence between a rare neurodegenerative disease of early life and a“common” neurodegenerative disease of late life has been observed before,e.g.for GD and PD.Here,we summarized links between AD and NCLs and we found accumulating overlap.
Several overlapping data are based on clinical observations.These are complex considering that other dementia syndromes can mimic AD at the time of the initial evaluation,there is yet no single behavioral marker that can reliably discriminate AD from other dementias and AD often co-occurs with other pathologies at autopsy (Karantzoulis and Galvin,2011).Apart from such diagnostic complexity,AD and NCLs present distinct clinical signatures.These may be explained in large parts by the different ages of onset.In NCLs,neurodegeneration occurs mostly in a still developing brain and not in a mature brain as in AD.The early neurodegeneration of neurons may cause several symptoms,such as frequently observed progressive epilepsies that are also found in other lysosomal childhood neurodegenerative diseases(Minassian,2014).Notably,patients with common AD will in the most advanced stages develop also myoclonus,and patients with the aggressive AD of Down syndrome develop a typical and severe progressive myoclonus epilepsy.However,the time point of manifestation clearly results in modified clinical signatures and different severity as observed in classicalversusprotracted forms of NCLs or early onsetversuslate onset AD.
As summarized above,several studies demonstrate an eti ologic and genetic overlap between NCLs and AD.Notably,the number of studies is still restricted and limits this review and our view on converging links between AD and NCLs.Extending the use of genetic disease diagnosis and genetic linkage analysis will be important to provide additional corroboration.
There is abundant evidence for dysfunction of the autophagy-endolysosome system contributing to the pathogenesis of both,AD and NCLs,and that this dysfunction is a major driver of neurodegeneration in both diseases.Different mutations and gene dosage effects are likely dictating the severity of dysfunction and the time point of manifestation in different subtypes.Loss of function or gradual residual functions may result in a phenotypic conti nuum ranging from severe and early onset to less severe,protracted,or late onset and even to asymptomatic cases.The severity of the diseases is likely also modulated by additional comorbidities and genetic risk factors.
The convergence of lysosomal and autophagy dysfunction could provide a foundation for the development of common therapeutic designs or the transfer of therapeutic strategies from one disease to the other.However,dysfunction of the autophagy-endolysosome system is not limited to NCLs and AD.Such dysfunction and progressive loss of nerve cells are commonly observed also in other neurodegenerative diseases,such as FTLD or PD.Strikingly,a distinct pattern of regional atrophy,neuronal degeneration and accumulation of specific material are characteristic pathological hallmarks of the individual disorders.Thus,lysosome and autophagosome dysfunction alone cannot explain the disease specific occurrence of neuronal vulnerability in each disease and understanding the specific underlying mechanisms is sti ll a challenging task.However,the development of targeted therapies that enhance or repair lysosome and autophagosome function in the developing and aged brain may have broad therapeutic utility and are a particularly promising area for future research.
Acknowledgments:The authors apologize to all colleagues whose important contributions to this field could not be cited due to space constrains.
Author contributions:MK and GH performed literature searches,outlinedand wrote the manuscript.MK created the figure.Both authors approved the final manuscript.
Conflicts of interest:No potenti al conflict of interest by the authors.
Open access statement:This is an open access journal,andarticles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Bystanders or not? Microglia and lymphocytes in aging and stroke
- Alzheimer’s disease risk after COVID-19: a view from the perspective of the infecti ous hypothesis of neurodegeneration
- Serine and arginine rich splicing factor 1: a potenti al target for neuroprotection and other diseases
- Can glial cells save neurons in epilepsy?
- Lights for epilepsy: can photobiomodulation reduce seizures and offer neuroprotection?
- The landscape of cognitive impairment in superoxide dismutase 1-amyotrophic lateral sclerosis