
Regulation of enolase activation to promote neural protection and regeneration in spinal cord injury
2023-02-24 05:24HannahMcCoyRachelPolcynNarenBanikAzizulHaque
中国神经再生研究(英文版) 2023年7期
Hannah M.McCoy ,Rachel Polcyn ,Naren L.Banik, ,Azizul Haque,
Abstract Spinal cord injury (SCI) is a debilitating condition characterized by damage to the spinal cord resulting in loss of function,mobility,and sensation with no U.S.Food and Drug Administration-approved cure.Enolase,a multifunctional glycolytic enzyme upregulated after SCI,promotes pro-and antiinflammatory events and regulates functional recovery in SCI.Enolase is normally expressed in the cytosol,but the expression is upregulated at the cell surface following cellular injury,promoting glial cell activation and signal transduction pathway activation.SCI-induced microglia activation triggers pro-inflammatory mediators at the injury site,activating other immune cells and metabolic events,i.e.,Rho-associated kinase,contributing to the neuroinflammation found in SCI.Enolase surface expression also activates cathepsin X,resulting in cleavage of the C-terminal end of neuron-specific enolase (NSE) and non-neuronal enolase (NNE).Fully functional enolase is necessary as NSE/NNE C-terminal proteins activate many neurotrophic processes,i.e.,the plasminogen activation system,phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B,and mitogen-activated protein kinase/extracellular signal-regulated kinase.Studies here suggest an enolase inhibitor,ENOblock,attenuates the activation of Rho-associated kinase,which may decrease glial cell activation and promote functional recovery following SCI.Also,ENOblock inhibits cathepsin X,which may help prevent the cleavage of the neurotrophic C-terminal protein allowing full plasminogen activation and phosphatidylinositol-4,5-bisphosphate 3-kinase/mitogen-activated protein kinase activity.The combined NSE/cathepsin X inhibition may serve as a potential therapeutic strategy for preventing neuroinflammation/degeneration and promoting neural cell regeneration and recovery following SCI.The role of cell membrane-expressed enolase and associated metabolic events should be investigated to determine if the same strategies can be applied to other neurodegenerative diseases.Hence,this review discusses the importance of enolase activation and inhibition as a potential therapeutic target following SCI to promote neuronal survival and regeneration.
Key Words:cathepsin X;ENOblock;enolase;glia;mitogen-activated protein kinase/extracellular signal-regulated kinase;neurodegeneration;neuroinflammation;phosphatidylinositol-4,5-bisphosphate 3-kinase/protein kinase B;Rho-associated protein kinase;spinal cord injury
Introduction
Spinal cord injury (SCI) is a devastating condition characterized by neuroinflammation and degeneration that can cause patients to lose their mobility,sensation,and the overall bodily function of their spinal cord(Polcyn et al.,2020).Primary SCI is the first initial insult to the spinal cord resulting in tissue damage at the site of the injury (Cox et al.,2015;Polcyn et al.,2017a,2020).Secondary SCI progressively develops following primary SCI through alterations of several cellular and metabolic events,resulting in increased neuroinflammation and neurodegeneration (Polcyn et al.,2017a).Primary SCI is irreversible,but secondary SCI,on the other hand,is reversible and is therefore viewed as an opportunity window for therapy to promote neuroregeneration and functional recovery following SCI (Tator and Fehlings,1991;Cox et al.,2015;Polcyn et al.,2020).Therapeutics targeting secondary injury mechanisms should be investigated to fully understand the connection between inflammation in SCI and the cells/mechanisms of action at work,i.e.,enolase.Enolase is a multi functional glycolytic enzyme,normally expressed in the cytosol,found to be upregulated on the cell surface following SCI in neural cells (Shimizu et al.,1983;Merkulova et al.,2000;Díaz-Ramos et al.,2012a;Haque et al.,2018).Surface-bound enolase assists in host cell invasion through plasminogen receptor binding,resulting in active plasmin generation,extracellular matrix degradation,and pathogen invasion as well as the activation of several neurotrophic signal transduction pathways(Haque et al.,2016,2017).Enolase activates cells of the immune system,including microglia and astrocytes,through the release of metabolic factors such as reactive oxygen species,nitric oxide,and different pro-and antiinflammatory cytokines and chemokines (Haque et al.,2016,2018;Polcyn et al.,2017a,b).Enolase also activates several metabolic pathways including the phosphati dylinositol-4,5-bisphosphate 3-kinase/protein kinase B (PI3K/AKT)and mitogen-activated protein kinase/extracellular signal-regulated kinase(MAPK/ERK) signal transduction pathways (Hafner et al.,2012;Zhang et al.,2015;Shi et al.,2019;Chung et al.,2020).These pathways are responsible for many effector functions,including neurotrophic activity and neuronal growth regulation as well as pro-inflammatory mechanisms (Yang et al.,2019;Chung et al.,2020).Taken together,these studies suggest enolase serves as an enzyme with both pro-and anti -inflammatory,giving reasons to believe enolase has different metabolic functions among its different isozymes.
Studies identified an isozyme of enolase,neuron-specific enolase (NSE)(an isoform/hybrid containing γ-enolase),that functions to promote neural repair and regeneration through its C-terminal protein,but also contributes to the inflammatory response (Kimura et al.,1994;López-Alemany et al.,2003;Obermajer et al.,2006,2009;Vizin and Kos,2015).Our recent study suggests enolase activation is detected after acute SCI as evidenced by the upregulation of NSE expression in rat serum and tissue (Polcyn et al.,2020).Our study determined a connection exists between increased NSE activity and pro-inflammatory events in SCI,i.e.,increased pro-inflammatory cytokine and chemokine levels and the alteration of key metabolic factors (Haque et al.,2016,2018;Polcyn et al.,2020).Through this study,our lab identified an enolase inhibitor,ENOblock,that attenuates pro-inflammatory activity following SCI through altering metabolic hormone levels associated with inflammation and the reduction of pro-inflammatory cytokines/chemokines Findings suggest ENOblock treatment reduces neuroinflammation associated with secondary mechanisms of SCI and favors anti-inflammatory effects.Hence,ENOblock may be used as a potenti al therapeutic strategy to improve functional outcomes and increase neuronal survival following SCI (Polcyn et al.,2020).
Studies also show NSE cell surface expression can promote neuroinflammation and degeneration through activation of the Rho-A/Rho-associated kinase(ROCK) pathway (Hashimoto et al.,1999;Hafner et al.,2012,2018;Polcyn et al.,2017b).Enolase activation of the PI3K/AKT and MAPK/ERK signaling pathways activates the ROCK pathway,a major regulator of acti n-cytoskeleton formation and organization as well as glial cell activation,proliferation,and differentiation (Hafner et al.,2012;Roser et al.,2017).Activation of the ROCK pathway is associated with increased levels of neuroinflammation and neurodegeneration,however,attenuation of the ROCK signaling pathway can lead to neuronal regeneration and survival following central nervous system(CNS) injury (Mueller et al.,2005;Haque et al.,2016;Jia et al.,2016).NSE surface acti vity also activates cathepsin X (Cat X),a cysteine carboxypepti dase enzyme,responsible for the regulation of immune and neuronal cells(Obermajer et al.,2006,2009;Hafner et al.,2012,2013;Fonović et al.,2017).Cat X is responsible for the cleavage of the C-terminus end of NSE,rendering NSE dysfunctional,resulting in neuroinflammation and degeneration(Obermajer et al.,2006,2009;Hafner et al.,2012,2013;Fonović et al.,2017).Inhibition of NSE is associated with decreased levels of Cat X,resulting in the attenuation of neuronal death (Obermajer et al.,2006;Fonović et al.,2017).
It is interesting to see how enolase functions as both a pro-survival and prodeath enzyme in the metabolic pathways implicated in SCI (Hattori et al.,1995;Mueller et al.,2005;Zhang et al.,2013;Haque et al.,2016,2017,2018;Roser et al.,2017;Shields et al.,2020).Investigations into enolase inhibition show the attenuation of neuroinflammation and degeneration and the promotion of neural repair and regeneration following SCI (Nakagawa et al.,1996;Mueller et al.,2005;Zhang et al.,2013;Haque et al.,2016,2017,2018;Polcyn et al.,2020).These studies show enolase inhibition prevents cleavage of the NSE C-terminal protein by Cat X inhibition,and also prevents Rho/ROCK pathway activation following SCI (Nakagawa et al.,1996;Mueller et al.,2005;Zhang et al.,2013;Haque et al.,2017,2018;Polcyn et al.,2020).However,the full outcome of enolase inhibition is unclear and should be further investigated to determine more potenti al functions of enolase inhibition.The importance of enolase activation in SCI and its inhibition as a potenti al target to promote neuronal regeneration and survival is discussed in this review.
Literature Search Strategy
PubMed was used as the main database to search for information as well as Web of Science and ScienceDirect.Dates of searching for data range from May 13,2021 to August 9,2022.The electronic search strategy,taking PubMed as an example,included searching key words such as ‘spinal cord injury’,‘enolase’,‘microglia’,‘cathepsin X’,etc.together and analyzing the results.Manuscripts including multiple key words were read through and relative information was noted from each manuscript.Manuscripts older than 10 years were screened for relevance.Manuscripts newer than 5 years were preferred as the information is more relevant to research present day.Manuscripts acknowledging the topic of discussion/containing keywords but not giving new insights or manuscripts repeating information were dismissed if not relevant.Manuscripts initi ally selected for use were screened out if the same information was found in more recently published manuscripts.Over 70 manuscripts were screened and 58 were selected to be included in the final manuscript based on the criteria described.
Spinal Cord Injury
SCI is a debilitating condition characterized by damage to the spinal cord resulting in loss of function,mobility,and sensation affecting approximately 282,000 people in the USA alone (Varma et al.,2013;National Spinal Cord Injury Stati stical Center,2018;Polcyn et al.,2020).Clinical trials investigating therapeutic options have been conducted in certain instances of acute and chronic SCI but have not shown clinical significance,making investigations into treatment options necessary (Varma et al.,2013;Cox et al.,2015).SCI progressively develops,starting with the first phase,or immediate primary injury,and the second phase,or secondary injury.Primary trauma is caused by external mechanical forces,such as lacerations,contusions,contractions,or compressions of the spinal cord,resulting in irreversible tissue damage at the impact site (Cox et al.,2015;Polcyn et al.,2017a,2020).Primary injuries can lead to severe necrosis of the injured tissue and are considered irreversible processes (Tator and Fehlings,1991;Cox et al.,2015;Polcyn et al.,2020).Secondary trauma,on the other hand,consists of several cellular and molecular events developing days to weeks following the primary injury(Polcyn et al.,2017a).
There are several mechanisms of action at work in secondary injury,ranging from acute to chronic mechanisms,including hypoxia and ischemia,glial scarring,gliosis,axonal damage,myelin loss,neuroinflammation,neuronal death,etc.that develop over time (Tator and Fehlings,1991;Cox et al.,2015).These secondary injury mechanisms are caused by a variety of cells including microglial,astrocytes,endothelial cells,Schwann cells,fibroblasts,etc.(Orr and Gensel,2018).Secondary trauma exacerbates neuroinflammation,degeneration,and cell death but also serves a regenerative capacity as well,making secondary trauma a reversible process.Glial cells,for example,not only potentiate SCI trauma through modification of the glial scar but also have the capacity to facilitate endogenous repair (Orr and Gensel,2018).Secondary injury,therefore,is viewed as providing an opportunity window to attenuate injury progression and promote recovery in SCI (Tator and Fehlings,1991;Cox et al.,2015;Polcyn et al.,2020).Therapeutics targeting secondary injury mechanisms should be thoroughly investigated to fully understand the connection between inflammation in SCI and the cells/mechanisms of action at work.For example,Enolase,a glycolytic enzyme up-regulated in SCI,activates several metabolic cascades resulting in a variety of inflammatory mechanisms,i.e.,Rho A activation and gliosis (Haque et al.,2016,2018;Miranpuri et al.,2021).Rho A,a protein responsible for the regulation of critical biological processes such as cell growth,transformation,apoptosis,etc.,is activated by surface enolase,resulting in inflammatory cascades that further contribute to neuronal death (Hafner et al.,2012;Haque et al.,2016,2018).The ROCK pathway,activated by Rho A,further activates glial cells such as microglia and astrocytes (Merkulova et al.,2000;Zhang et al.,2013;Haque et al.,2016,2018;Miranpuri et al.,2021).Microglia undergo differenti ation to the M1 phenotype,resulting in pro-inflammatory cytokine and chemokine production as well as other pro-inflammatory events (Merkulova et al.,2000;Hu et al.,2015;Haque et al.,2018).Gliosis occurs following SCI and results in the proliferation and hypertrophy of astrocytes,ultimately contributing to neurodegeneration in the CNS through the release of pro-inflammatory cytokines and chemokines (Haque et al.,2018;Okada et al.,2018;Yang et al.,2019).These processes are interconnected and further contribute to SCI through interaction together,hence,therapeutics targeting several secondary injury mechanisms are necessary for the survival of neurons following SCI.
Multi functional Enolase
A multifunctional glycolytic enzyme,enolase,has been identified as a promising therapeutic target following SCI (Haque et al.,2016).Enolase is abundantly expressed in the cytosol and catalyzes the dehydration of 2-phosphoglycerate to phosphoenolpyruvate during glycolysis (Shimizu et al.,1983;Merkulova et al.,2000;Haque et al.,2018).Enolase also acts as a plasminogen binding protein and plays several roles in the immune response including growth control,immune activation,inflammation,and allergic responses (Haque et al.,2017).The expression of enolase on the surface of different cell types depends on the pathophysiological conditions of the cells (Díaz-Ramos et al.,2012a).Some studies have speculated inflammatory stimulation of monocytes and microglia induces the rapid translocation of enolase to the cell surface,as well as other inflammatory sti muli and growth factors,but the molecular mechanism driving enolase translocation to the cell surface is not fully understood (Wygrecka et al.,2009;Bae et al.,2012;Didiasova et al.,2019).Enolase can cross the plasma membrane into the extracellular space in the form of exosomes or as a soluble protein (Didiasova et al.,2019).The surface-bound and secreted enolase proteins both display strong plasminogen binding capacity and alter extracellular proteolytic acti vity (Didiasova et al.,2019).Surface-bound enolase interacts at the cell surface,providing enhanced anti gen presentation for host cell invasions and degradation of the extracellular matrix (Polcyn et al.,2017a).Enolase cell surface expression also triggers reactive oxygen species,nitric oxide,and proinflammatory cytokines and chemokines,making enolase a contributor to inflammation pathogenesis (Hafner et al.,2012;Haque et al.,2016;Polcyn et al.,2017a).Enolase cell surface expression also contributes to neuronal growth and survival as enolase activates the plasminogen system,producing plasmin and activating many neuroprotective signaling pathways (Butterfield and Lange,2009;Díaz-Ramos et al.,2012a;Sawhney et al.,2015).The exact mechanisms by which enolase acts are still being investigated,however,different types of enolase isozymes have been identified that serve different purposes and are present in different tissues in the body.
Three tissue-specific subunits of enolases exist α-enolase,β-enolase,and γ-enolase.α-Enolase is ubiquitous,β-enolase is muscle-specific,and γ-enolase is neuron-specific (Shimizu et al.,1983;Merkulova et al.,2000;Haque et al.,2018).α-Enolase can convert to either β-enolase and/or γ-enolase during injury,disease,or natural development.Sometimes,isotype switching is not always successful,and a hybrid dimeric enolase isozyme is formed,αγ-enolase for example (Shimizu et al.,1983;Merkulova et al.,2000;Haque et al.,2018).Other enolase isozymes include αα-enolase,αβ-enolase,ββ-enolase,and γγenolase.The γγ-enolase isozyme is present in neurons,oligodendrocytes,and microglia,and the αγ-enolase hybrid enzyme is present in neurons,microglia,and astrocytes (Kimura et al.,1994;Obermajer et al.,2009;Haque et al.,2018).Studies have shown following SCI,several enolase isozymes are present at elevated levels in astrocytes,microglia,and neurons (Haque et al.,2018).As previously mentioned,enolase is believed to migrate to the cell surface following injury,giving reason to believe following SCI,enolase isozymes are present on the cell surface of neuronal cells (Haque et al.,2016).α-Enolase contains a C-terminal lysine residue that allows the protein to bind plasminogen on the cell surface,giving enolase some neuroprotective characteristics (Hafner et al.,2012;Vizin and Kos,2015).In contrast,γ-enolase does not contain a C-terminal lysine residue,but recent studies have speculated γ-enolase contains a hydrophobic domain in the N-terminal region that allows the protein to associate with the plasma membrane and other cell surfaces (Vizin and Kos,2015).These mechanisms are believed to be at work following SCI,indicated by the presence of NSE at elevated levels in neuronal cells.
In neurodegenerative diseases,the extent of neural cell loss correlates with increased NSE levels in serum and cerebrospinal fluid as well as with the clinical progression of neurodegenerative diseases (Polcyn et al.,2017b;Haque et al.,2018;Tsukahara et al.,2021).Studies have shown elevated NSE levels in the cerebrospinal fluid of rodent models after injection with kainic acid,a receptor agonist responsible for inducing excitotoxic neuron death,and other animal models of SCI (Ondruschka et al.,2013;Haque et al.,2018).Studies have also shown elevated NSE levels in the grey matter of the adult brain following traumatic brain injury (TBI),indicating NSE may serve as a potenti al marker of lesions in the brain (Ondruschka et al.,2013;Haque et al.,2018).These interactions of NSE with different cells and molecules of the CNS suggest NSE is involved in neuroinflammation,glial cell activation,and neuronal damage but the exact mechanisms by which NSE interacts remain unclear (Polcyn et al.,2017b).These results also give one reason to believe NSE can be used as a potenti al biomarker of functional damage to neurons in SCI and other neurodegenerative diseases (Haque et al.,2018;Tsukahara et al.,2021).NSE promotes extracellular degradation,inflammatory glial cell proliferation,and actin remodeling,promotes the migration of activated microglia to injury sites,and increases neuronal cell death (Haque et al.,2018).Elevated levels of NSE following SCI may also stimulate the production of reactive oxygen species,nitric oxide,and pro-inflammatory cytokines and chemokines,inducing apoptosis of neural cells (Haque et al.,2016,2017).NSE pro-inflammatory acti vity is necessary for maintaining normal homeostasis,but excessive inflammatory responses can lead to further neuroinflammation and neurodegeneration.
Studies have also shown NSE to have a neurotrophic capacity and is essenti al for the survival of neurons following SCI (Hafner et al.,2013;Vizin and Kos,2015;Haque et al.,2016,2018).NSE is involved in neural cell repair following SCI and is considered neurotrophic as NSE controls neuronal differenti ation,neurite regeneration,and overall neuronal survival (Hafner et al.,2012;Haque et al.,2018).The C-terminal protein of NSE controls neural survival,differentiation,and neurogenesis by activating the PI3K/AKT and MAPK/ERK signal transduction pathways (Polcyn et al.,2017b;Haque et al.,2018;Shi et al.,2019;Guo et al.,2020).The PI3K/AKT pathway is an intracellular signal transduction pathway responsible for promoting metabolism,cell proliferation,growth,and survival in response to extracellular signals,i.e.,serine and/or threonine phosphorylation cascades (Zhang et al.,2015;Yang et al.,2019).MAPK/ERK signaling pathway is also responsible for relaying,amplifying,and integrating extracellular signals to elicit physiologic responses such as cell differentiation,proliferation,development,inflammatory responses,and apoptosis (Guo et al.,2020).Activation of the PI3K/AKT and MAPK/ERK signal pathways is required for neurotrophic activity and growth regulation but can also contribute to neuroinflammation due to proinflammatory effector functions (Zhang et al.,2015;Chung et al.,2020).The signaling pathways are responsible for the increased expression of a specific growth-associated protein,GAP-43 (Hafner et al.,2012;Chung et al.,2020).PI3K/AKT and MAPK/ERK are also responsible for the regulation of Rho A kinase,a key regulator of acti n-cytoskeleton formation and organization,and the ROCK signaling pathway (Hafner et al.,2012,2018).These factors may give NSE its multi functional role in neuroprotection and neurodegeneration(Hattori et al.,1995),but the full mechanism of the C-terminus activity of NSE remains unclear and further analysis needs to be conducted to fully understand NSE neurotrophic acti vity.
Upregulation of Cell Surface α-Enolase Triggers Inflammatory Cycles
As previously mentioned,studies have shown enolase migrates from the cytosol to the cell surface following stimulation by various metabolic factors (Butterfield and Lange,2009;Bae et al.,2012;Sawhney et al.,2015).α-Enolase,the most abundant enolase isoform,carries out its normal metabolic role in the cytoplasm but moves to the cell surface by an unknown mechanism (Butterfield and Lange,2009;Sawhney et al.,2015).It is speculated a hydrophobic domain within α-enolase exists that serves as an internal signal sequence,allowing its integration into the cell membrane while retaining its enzymatic acti vity (Butterfield and Lange,2009).Other studies speculate post-translational acylation or phosphorylation occurs,allowing the integration,however,no clear mechanism has been identified (Butterfield and Lange,2009).α-Enolase cell surface expression allows the enzyme to function as a plasminogen surface receptor,activating the plasminogen activator(PA) system in the presence of either tissue PA (tPA) or urokinase PA (uPA)(Butterfield and Lange,2009;Díaz-Ramos et al.,2012b;Sawhney et al.,2015;Whyte and Mutch,2021).Both tPA and uPA are PAs normally found in the CNS and are responsible for the cleavage of plasminogen into plasmin in the PA system (Butterfield and Lange,2009;Díaz-Ramos et al.,2012b).
The PA system is an enzymatic cascade involved in the regulation and acti vity of plasmin that also plays a key role in maintaining normal homeostasis,thrombosis,and other biological processes (Bharadwaj et al.,2021).The PA system is implicated in wound healing and inflammation through its involvement in cell migration and proliferation as well as intracellular signaling events through the activation of pro-enzymes,hormones,growth factors,and cytokines (Butterfield and Lange,2009;Sawhney et al.,2015;Napolitano and Montuori,2021).The PA system localizes monocytes and microglia to specific areas of the CNS where damaged neurons are located,as seen in patients with Alzheimer’s disease and rheumatoid arthritis (Butterfield and Lange,2009;Bae et al.,2012).It is speculated similar mechanisms of action are at work in patients with SCI,indicating the need for further research in other neurodegenerative disease models.Studies have shown the PA system’s effects are more disastrous in the presence of α-enolase on neuronal cell surfaces (Butterfield and Lange,2009).The binding of plasminogen to α-enolase rapidly activates the PA system through tPA/uPA proteolytic cleavage (Butterfield and Lange,2009;Sawhney et al.,2015).The binding of plasminogen to enolase also prevents inhibition of the PA system by blocking plasminogen inhibitor binding (Butterfield and Lange,2009).The activated PA system induces plasmin-mediated lysis as well as the activation of several inflammatory pathways.The proteolytic acti vity of plasmin activates MAPK/ERK,as well as other pro-inflammatory pathways,i.e.,nuclear factor-κB,directly leading to the transcriptional regulation of α-enolase (Butterfield and Lange,2009;Bae et al.,2012;Sawhney et al.,2015).Studies have shown,in patients with Alzheimer’s disease,MAPK/ERK is active in areas of the brain associated with neuronal damage,indicating similar processes may be at work in SCI (Butterfield and Lange,2009).The exact mechanism by which MAPK regulates α-enolase is unknown,however,some studies speculate MAPK/ERK may induce α-enolase expression throughc-Mycpromoter binding (Butterfield and Lange,2009).c-Mycis a proto-oncogene that serves as a master regulator of cellular metabolism and proliferation.
Studies also speculate that MAPK/ERK regulation of α-enolase occurs through the hypoxia-inducible factor 1,a transcription factor,that plays an integral role in the body’s response to hypoxia (Butterfield and Lange,2009).Studies have shown bothc-Mycand hypoxia-inducible factor 1 may up-regulate glucose metabolism via ERK signaling under hyper-metabolic conditions,i.e.,Alzheimer’s disease and other neurodegenerative diseases (Butterfield and Lange,2009).These results give one reason to believe under hypermetabolic conditions such as oxidative stress,hypoxia,etc.,activate neuronal and glial intracellular survival pathways (MAPK/ERK),leading to increased enolase levels in the CNS and a never-ending,perpetual cycle (Butterfield and Lange,2009;Bae et al.,2012).Enolase-bound plasminogen,when activated by tPA/uPA,stimulates plasmin activation of MAPK/ERK signaling cascades that in turn up-regulate the transcription of glycolytic enzymes to counteract the hyper-metabolic environment and prevent apoptosis (Butterfield and Lange,2009;Bae et al.,2012;Sawhney et al.,2015).Thus,MAPK/ERK signaling pathways can function in a neuroprotective manner through upregulation of enolase,however,hyper-activation of MAPK/ERK may render the CNS sensitive to neurodegeneration as MAPK/ERK activates many proinflammatory cytokines (tumor necrosis factor-α,interferon-γ,prostaglandin E,etc.) and signaling cascades,i.e.,ROCK (Bae et al.,2012).Increased enolase levels may also result in an increase in anti bodies targeted against enolase,aka enolase autoantibodies (Bae et al.,2012).Enolase autoantibodies bind to enolase,resulting in increases in pro-inflammatory cytokine production by monocytes and microglia,and further enolase migration to the cell surface(Bae et al.,2012).The cycle conti nues with increases in plasmin generation,providing cells with enhanced migratory and invasive properties (Bae et al.,2012;Sawhney et al.,2015).Hence,enolase cell surface expression can result in both pro-inflammatory/neurodegenerative and anti -inflammatory/neuroprotective processes.
Localization of NSE in neural cells demonstrates NSE functions as both a proinflammatory and anti-inflammatory or neurotrophic enzyme,regulating neural growth,differenti ation,and survival (Haque et al.,2018).Both pro-and anti -inflammatory mechanisms of action of NSE occur because of its presence not only in neural cells,but also through interactions with glial cells such as microglia,astrocytes,and oligodendrocytes through the activation of several metabolic pathways,i.e.,PI3K/AKT,MAPK/ERK,and ROCK pathways (Haque et al.,2018).
Glial Cell Activation by Rho-Associated Protein Kinase Pathway
ROCK is a serine/threonine kinase expressed as two homologs,ROCK I and ROCK II,known to activate glial cells following activation by NSE in SCI(Nakagawa et al.,1996;Roser et al.,2017).Both homologs share a similar structure and function but are present in different locations throughout the body.ROCK I is the predominant isoform in the liver,lungs,testi s,blood,and immune system,and ROCK II is the predominant isoform in the brain and muscles (Nakagawa et al.,1996;Hashimoto et al.,1999;Roser et al.,2017).An upstream regulator from the Ras-superfamily,Rho-GTPase RhoA is a key regulator of ROCK (Hashimoto et al.,1999;Roser et al.,2017).The ROCK pathway is involved in the regulation of microglia migration and phagocytosis,as well as in the release of inflammatory cytokines and chemokines,and consequently affects microglia phenotype (Mueller et al.,2005;Roser et al.,2017).
Microglial precursor cells differentiate into either M1 or M2 microglia following activation by ROCK.M1 microglia are cytotoxic and contribute to neurodegeneration and neural cell death following SCI (Zhang et al.,2013;Haque et al.,2018;Shields et al.,2020).Oppositely,M2 microglia are neuroprotective and contribute to the repair process by producing neurotrophic factors following SCI (Zhang et al.,2013;Haque et al.,2018;Shields et al.,2020).Pro-inflammatory microglia (M1),activated by ROCK,release cytokines (interleukin-1α,tumor necrosis factor-α,complement component 1q) resulting in neuronal cell death through several mechanisms including astrocyte differentiation into dysfunctional phenotypes (Liddelow et al.,2017).Astrocytes are the most numerous cells in the CNS responsible for a variety of functions,including regulatory roles in neurogenesis,synaptogenesis,blood-brain barrier maintenance,and extracellular homeostasis (Siracusa et al.,2019).Analogous to microglia differenti ation,astrocytes differenti ate into A1 or A2 astrocytes,Induction of the A1 phenotype causes astrocytes to lose the ability to promote neuronal growth,synaptogenesis,phagocytosis,and overall neuronal survival whereas the A2 phenotype is neuroprotective,contributing to neuronal repair and regeneration (Liddelow et al.,2017).Studies have shown following SCI in animal models,levels of M1 microglia and A1 astrocytes are elevated in comparison to control as well as an increased number of M1 microglia and A1 astrocytes in comparison to M2 microglia and A2 astrocyte levels,respectively(Jiang et al.,2020;Li et al.,2020;Liu et al.,2022).There is speculation as to whether A1 astrocytes are able to revert to A2 astrocytes following M1 microglial activation in SCI.Investigations into glial cell acti vity following SCI need to be further investigated to understand the full therapeutic potenti al of M2 microglia and A2 astrocytes.
ROCK-associated microglial pro-inflammatory acti vity is necessary following SCI;however,hyper-activity of microglia augments neuroinflammation and neurodegeneration.Studies have shown inhibition of NSE may result in the attenuation of the RhoA/ROCK activation pathways and neuronal survival and regeneration (Haque et al.,2016).The exact mechanisms by which NSE inhibition contributes to neurogenesis,regeneration,and survival are unknown.Study results give reason to believe NSE/ROCK inhibition results in decreased inflammation and increased myelination,axonal protection,neurite outgrowth,tissue preservation,and regeneration (Mueller et al.,2005;Jia et al.,2016).Various studies have confirmed the beneficial features of ROCK inhibitors but more research needs to be conducted to determine the efficacy and safety of ROCK inhibitors before clinical use in humans (Mueller et al.,2005).
Both pro-and anti-inflammatory processes are necessary following SCI,giving reason to investigate therapeutic targets involved in both mechanisms,i.e.,NSE specifically.The relation between NSE cell surface expression and the RhoA/ROCK activation pathway suggests a potential therapeutic strategy following SCI to decrease neuroinflammation and degeneration as a decrease in cell surface NSE will attenuate the Rho/ROCK activation pathway.It is important to reduce pro-inflammatory factors following SCI without completely diminishing the inflammatory response.Pro-inflammatory mechanisms are essenti al to prevent further infection and maintain normal homeostasis following CNS injury,however,an abundance of inflammatory mechanisms can result in further neuroinflammation and degeneration(Kinney et al.,2018).A therapeutic technique permitting both pro-and anti -inflammatory mechanisms is speculated to produce the most significant clinical effects for recovery following SCI.
Cathepsin X and Cleavage of Enolase
Enolase’s presence on the cell surface has also been linked to the regulation of neural survival,differentiation,and regeneration following SCI by the cysteine carboxypepti dase enzyme,Cat X (Obermajer et al.,2009;Hafner et al.,2012;Fonović et al.,2017;Haque et al.,2018).Cat X is normally present in the perinuclear endo-lysosomal compartments and does not co-localize with enolase (Obermajer et al.,2006,2009).However,in differenti ated monocytes,translocation to the plasma membrane can occur and Cat X is able to interact with metabolic factors on the cell surface,i.e.,enolase (Obermajer et al.,2006,2009).Studies have shown the expression and localization of Cat X on the plasma membrane are increased with stimulation by neurotrophic growth factor,giving reason to believe Cat X may be stimulated by the neurotrophic aspects of NSE (Obermajer et al.,2006,2009).Cat X cleaves the C-terminal end of membrane-bound NSE and non-neuronal enolase,impairing enolase activity by abolishing both isozyme’s neurotrophic activity (Obermajer et al.,2009;Hafner et al.,2013;Fonović et al.,2017;Haque et al.,2018).Cat X cleavage of α-enolase (non-neuronal enolase) C-terminus results in dysfunction of plasminogen receptor binding (López-Alemany et al.,2003;Obermajer et al.,2009).Cleavage of γ-enolase (NSE) C-terminus results in dysregulation of early-stage neurogenesis and neuron survival due to the loss of an additional active site in the C-terminus responsible for the activation of pathways promoting neuron survival and neurogenesis (Kimura et al.,1994;Obermajer et al.,2009;Hafner et al.,2012,2013;Fonović et al.,2017).
Increased levels of Cat X contribute to the attenuation of NSE expression and activity in injured tissues (Haque et al.,2018).These results suggest Cat X acti vity might contribute to neurodegeneration following SCI by impairment of NSE’s neurotrophic aspects (Haque et al.,2016,2018).Hence,overexpression of Cat X can be detrimental because the level of NSE required for neural cell survival is lost as a result of the catalytic acti vity of Cat X.Inhibition of Cat X activity may promote cell survival and cell differentiation due to the neurotrophic activity of the C-terminal end of both α-and γ-enolases on the cell surface (Haque et al.,2018).The addition of the C-terminal end of NSE exerts a pro-survival role and changes the neurite length distribution pattern (Obermajer et al.,2009).Inhibition of NSE inflammatory activity decreases neuroinflammation and degeneration,however,it is important to not completely inhibit NSE to preserve the pro-survival aspects for neuronal cells (Obermajer et al.,2009).A combination approach targeting both NSE and Cat X should be investigated to achieve parti al inhibition of the pro-death aspects of NSE to support the pro-survival aspects of NSE in decreasing neural degeneration and increasing neural regeneration and survival.
Regulation of Neuron-Specific Enolase and Cathepsin X
Recent studies have shown NSE inhibition to be a potential treatment strategy for SCI (Haque et al.,2016;Polcyn et al.,2020).ENOblock,an enolase inhibitor,alters inflammatory metabolic factors,chemokines,and cytokines following SCI in rat contusion models (Haque et al.,2018).Attenuation of the Rho/ROCK signaling pathway by NSE inhibition decreases the inflammatory response due to decreased glial cell activation and differenti ation into proinflammatory phenotypes (Nakagawa et al.,1996;Mueller et al.,2005;Zhang et al.,2013;Haque et al.,2018).Ongoing studies in our laboratory suggest the inhibition of NSE by ENOblock attenuates Rho A/ROCK II in BV-2 microglia cell lines (Figure 1A),which in turn may inhibit M1 microglia/A1 astrocytes and attenuate inflammation and neurodegeneration,as well as promote repair by M2 microglia/A2 astrocytes.The exact mechanism by which ENOblock inhibits NSE is unknown,but there is reason to believe ENOblock inhibits the prodeath aspects of enolase,giving room for the pro-survival aspects.
Studies performed in our laboratory also show that NSE inhibition by ENOblock decreases Cat X expression in VSC4.1 motoneuron cells (Figure 1B).Enolase inhibition also protects neuronal cells (Figure 1C).These unpublished data support in favor of earlier findings that decreased Cat X levels may prevent cleavage of the C-terminal end of NSE,resulting in neural survival and axonal protection.Overall,ENOblock decreases neurodegeneration and improves neural survival and protection of axons by the inhibition of NSE and Cat X activation following SCI as shown inFigure 2.The combined aspects of ENOblock inhibition of enolase/NSE activation produce a perfect environment for neural regeneration to occur.Enolase inhibition attenuates the ROCK activation pathway and inhibits Cat X cleavage of NSE as shown in the diagram(Figure 2AandB),leading to neuron regeneration and survival.
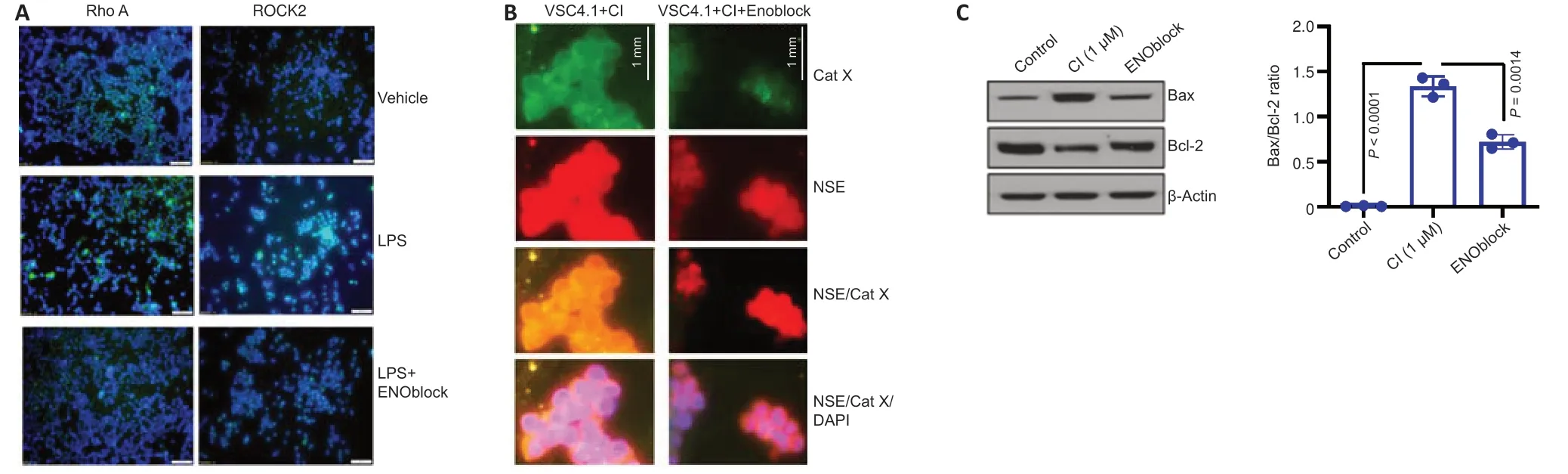
Figure 1|Regulation of enolase activation may prevent neurodegeneration.
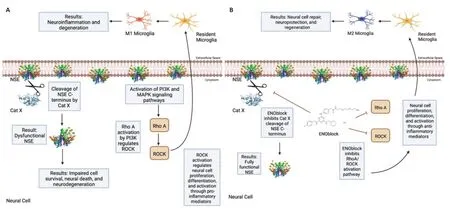
Figure 2|Inhibition of enolase activation may support neural regeneration.
ENOblock inhibition of NSE may lead to neuronal survival through additional metabolic events and factors.ENOblock inhibition of NSE has also resulted in a decrease in Ionized calcium-binding adapter molecule 1 and glial fibrillary acidic protein levels,decreasing microglia and astrocyte activation,respectively,and pro-inflammatory events following SCI in rat models(Haque et al.,2017;Polcyn et al.,2020).There has also been a decrease in cyclooxygenase 2 expression associated with NSE inhibition by ENOblock.Cyclooxygenase 2 is an enzyme essenti al for the synthesis of prostaglandins,which have a strong tendency to induce inflammation and cause symptoms of pain (Rawat et al.,2019).Enolase inhibition also increases myelin basic protein and neurofilament protein levels (Polcyn et al.,2020).Myelin basic protein elevation results in improved myelination and protection of axons and neurons.Elevated neurofilament protein levels are also associated with neural survival and axonal protection.ENOblock treatment attenuates the decrease in the neuronal cytoskeleton that occurs following SCI as evidenced by increased neurofilament protein levels in recovering neuronal cytoskeletons.ENOblock treatment in rodent SCI models is also shown to increase both insulin and glucagon levels,two hormones known to promote antiinflammatory activity in the CNS.Increased levels of insulin in the CNS promote anti -inflammatory effects and lead to neuroprotection through the regulation of nuclear factor-κB levels and interaction with neural cells,i.e.,microglia and astrocytes (Polcyn et al.,2020).Increased levels of glucagon decrease Th2 cytokines,tumor necrosis factor-α,and natural killer cell acti vity while increasing intracellular cyclic adenosine monophosphate levels (Polcyn et al.,2020).ENOblock treatment also reduces gliosis through attenuation of both microglial and astrocyte acti vity (Haque et al.,2018;Polcyn et al.,2020).These aspects of enolase inhibition,as well as the neurotrophic aspects of fully functional NSE C-terminal activity,are essential for neuron survival following SCI indicating there is a need to both inhibit and promote NSE acti vity.
Conclusions
This review summarizes the important metabolic pathways involved in SCI as well as the importance of enolase activity on the cell surface following SCI.Interaction at the cell surface allows enolase to contribute to neuroinflammation and degeneration through the immune cell activation,the production of pro-inflammatory mediators,and the activation of cell signaling pathways.This review also highlights NSE’s pro-survival aspects and shows NSE has the potenti al to serve as a neurotrophic enzyme.Enolase,therefore,is a unique enzyme contributing to neuro-degeneration after overactivation and regeneration at normal physiological levels simultaneously.Inhibition of certain aspects of NSE will allow the necessary pro-inflammatory mechanisms to occur while simultaneously suppressing negative pro-inflammatory mechanisms and promoting anti -inflammatory events.Enolase inhibition by ENOblock could also prevent NSE C-terminus cleavage by Cat X,giving rise to a fully functional,neurotrophic NSE.Thus,the role of enolase inhibition by ENOblock may serve as a potential therapeutic to not only inhibit neuroinflammation and neuronal death but to promote neural survival.The success of this strategy in preclinical models of SCI gives hope for successful alleviation of the more detrimental effects of SCI in human clinical trials.
Enolase activation may also occur in TBI and trigger inflammatory responses and degeneration in the CNS.Thus,inhibiting enolase activation could help reduce neuroinflammation and degeneration and improve functional outcomes in TBI and other neurodegenerative diseases such as Alzheimer’s disease,Parkinson’s disease,and multiple sclerosis.Investigations into combination strategies targeting several metabolic events following SCI and TBI should be conducted to fully understand the therapeutic potential.A combined therapeutic would be especially beneficial for combat veterans returning from war due to their suscepti bility to TBI and/or SCI.As the field continues to progress,a safe,rational,combination treatment strategy will hopefully be available for individuals suffering from SCI,TBI,and other neurodegenerative/inflammatory diseases.Information on enolase in neuroinflammation and neurodegeneration is sti ll an underexplored area of investigation,thus limiting the content of this review.Limited information is available on the role of enolase in neurodegeneration in SCI.Thus,some unpublished data from the Haque lab were included in this review.There is also limited information on the mechanisms by which enolase acts on the immune system following neuroinflammation.Additional research is necessary to understand enolase acti vity and impact on SCI,as well as for strategic use in other neurodegenerative diseases.
Author contributions:HMM reviewed literature,drew the figures,analyzed data,wrote and edited the manuscript.RP drew figure 1.NLB reviewedliterature and edited the manuscript.AH reviewed literature,designed overall content of the manuscript,and edited the manuscript.All the authors read and approved the final version of the manuscript.
Conflicts of interest:The author declares no conflict of interest.
Open access statement:This is an open access journal,andarticles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Bystanders or not? Microglia and lymphocytes in aging and stroke
- Alzheimer’s disease risk after COVID-19: a view from the perspective of the infecti ous hypothesis of neurodegeneration
- Serine and arginine rich splicing factor 1: a potenti al target for neuroprotection and other diseases
- Can glial cells save neurons in epilepsy?
- Lights for epilepsy: can photobiomodulation reduce seizures and offer neuroprotection?
- The landscape of cognitive impairment in superoxide dismutase 1-amyotrophic lateral sclerosis