
氧化呋咱合成策略、反应机理及其在含能材料研发中的应用
2023-02-22 02:52张俊林折巍青王伯周
含能材料 2023年2期
张俊林,周 静,2,折巍青,王伯周
(1. 西安近代化学研究所, 陕西 西安 710065; 2. 北京理工大学化学与化工学院, 北京 102488)
0 引 言
含能材料的主要研究目标之一是提升含能化合物能量与密度[1]。结构上,含能化合物通常由分子骨架与以硝基及其衍生化的官能团为主的致爆基团组成,其传统分子骨架以碳基骨架为主,适当刺激下,致爆基团与作为燃料部分的碳基骨架直接发生分子内氧化还原反应,生成碳氧化物及氮氧化物,释放大量的热量、对外做功[2-3]。为进一步突破含能材料高能化发展的瓶颈,高氮氧含量的杂环骨架,因其致密的分子结构和突出的生成焓,逐步取代传统的碳基骨架而广泛应用含能材料的研发[4-5]。
高性能氮杂骨架构建的重要途径是在氮杂骨架中引入N-氧化物单元,该方法不仅有利于形成密集的晶体堆积结构、提升骨架密度,同时骨架生成焓因氧含量的增加而得以提升[6-8]。氧化呋咱是典型的含N-氧化物单元的平面型氮杂骨架,具有致密的堆积结构,因其独特的氮氧原子排布在氧化呋咱骨架内部嵌入了一个“潜硝基”片段,使其氮氧键的强度显著弱于普通硝基中氮氧键的强度,但它的存在使氧化呋咱成为蕴含致爆基团结构的分子骨架,赋予了其在含能材料研发中独特的价值[9-12]。从结构角度上看,N-氧化物片段中,N—O 成键电子来源于氮原子上的孤对电子,氮和氧分别具有正电荷和负电荷,这种电荷分布导致氧化呋咱结构中连续氧-氮-氧-氮片段具有显著的互变异构倾向,并且这种互变异构过程多需通过双亚硝基型中间体。由于在加热状态氧化呋咱容易经过双亚硝基中间体以开环再闭环的过程形成互变异构体,因而氧化呋咱普遍热稳定性较差。而这种极其特殊的密集氮氧原子排布、较强的互变异构倾向和较差的热稳定性也大幅增加了氧化呋咱结构的合成难度[13-15]。
从合成角度上看,氧化呋咱合成的最大难点在于设计具有较强环化倾向的合成前体,令其在指定的条件下自发转化为蕴含氧-氮-氧-氮排布的氧化呋咱环系。这类转化往往需要较强的热力学驱动力,因而氧化呋咱一般通过缩合反应或氧化反应等实现,在形成骨架环系的同时释放高度稳定的小分子结构,如氮气、水、氯化氢等。迄今氧化呋咱合成方法主要包括[16]基于一般性缩合反应的方法,如邻硝基叠氮基脱氮缩合法、1,2-二肟氧化缩合法、邻硝基氨基氧化缩合法,以及基于1,3-偶极子环加成反应的缩合方法,如含氯肟碱性缩合法、偕二硝甲基/乙酸基缩合法等。此外,一些较为特殊的烯烃加成转化法、基于氯肟或肟的衍生化环化法等也可用于氧化呋咱结构的制备。本文就氧化呋咱合成方法进行综述和比较,讨论了相关的环化机理并依据不同的应用背景,总结了基于氧化呋咱骨架的含能化合物相关研究进展。
1 氧化呋咱合成策略与机理
1.1 分子内缩合环化法
1.1.1 邻硝基叠氮基缩合环化法
硝基与叠氮基是使用最广泛的致爆基团[14-15],利用相邻的硝基与叠氮基在加热或光照条件下脱除一分子氮气形成氧化呋咱骨架的合成方法无需加入外来试剂。从反应机理角度来看,Rauhut 等[17]认为邻硝基叠氮基脱氮缩合法是以硝基中氧原子进攻叠氮基启动,并通过平面过渡态的单步协同转化机制形成氧化呋咱结构,而这种机理推测与实验监测研究所得数据最为符合;在相关平面过渡态中,硝基的1 个氧原子和叠氮基之间已经具有较强的相互键合作用。而Kawano等[18]则认为在光激发条件下该转化机理有可能通过先脱除氮气形成三线态或单线态的乃春(氮烯)中间体实现,但作者未能成功捕获该高活性中间体,仅通过计算实验进行了探讨(Scheme 1)。在合成中,邻硝基叠氮基脱氮缩合法一般采用先引入硝基再引入叠氮基的策略,尤其当硝基邻位含易离去基团时,硝基的存在极大地增强了芳环所连易离去基团与叠氮基发生亲电取代反应的倾向。由于邻硝基叠氮基脱氮缩合法无需外来试剂,邻硝基叠氮基结构本身又具有较高的感度和爆炸风险,因此催生出“一锅法”合成法,反应体系中叠氮基亲电取代反应和与硝基的缩合环化反应一并完成[19](Scheme 1)。
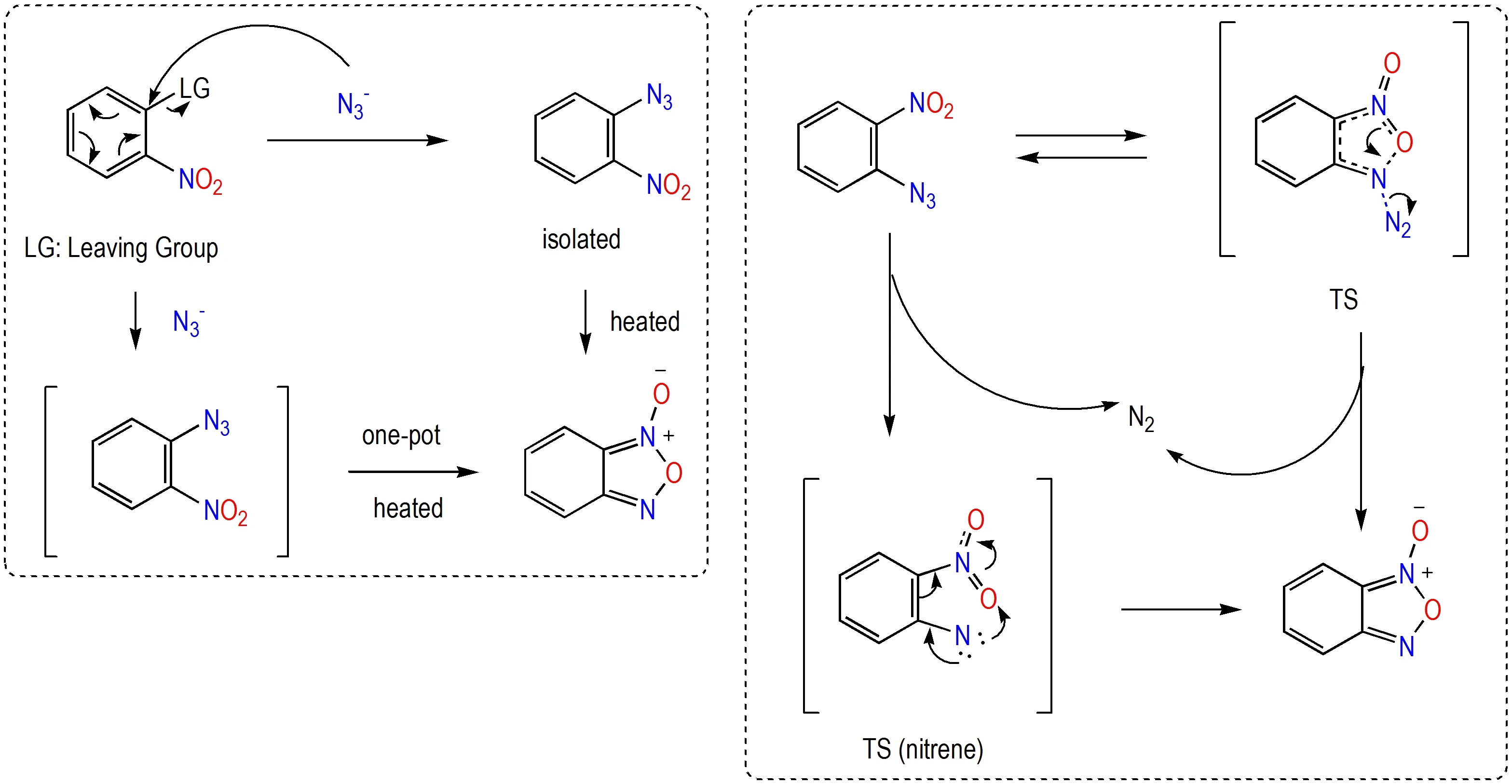
Scheme 1 Denitrogenation condensation of o-nitroazide structure and the mechanism[17-19]
1.1.2 1,2-二肟氧化缩合环化法
1,2-二肟氧化缩合法是氧化呋咱合成的常用方法,该方法由于先需要形成相邻的二肟结构,因此多基于易于获得1,2-二醛或1,2-二酮的底物。从总反应效果来看,1,2-二肟氧化缩合形成为氧化呋咱的过程共涉及2 个电子和2 个质子的转移,可选择的氧化剂种类很多,例如卤素、次卤酸盐、铁氰化物、高价铈离子、硝酸和氮氧化物、二氧化锰、四乙酸铅、N-碘代琥珀酰亚胺和苯基碘(Ⅲ)双三氟乙酸盐等,甚至直接以电化学氧化实现[20-22]。但对于该氧化环化反应的机理尚不完全明确,在电化学氧化过程中被认为可能是连续的单电子氧化机理,即首先为1,2-二肟经单电子氧化形成亚胺氧基自由基,然后进一步经单电子氧化形成氧代亚胺阳离子,最终经双电子氧化环化形成氧化呋咱[23]。Paine 等[24]利用实验监测手段探讨了双电子氧化机理,该在1,2-二肟氧化缩合过程中双电子氧化过程涉及到了氧化亚胺阳离子,而氧化亚胺阳离子是中性二亚硝基烯烃的互变异构形式,可环化形成氧化呋咱(Scheme 2)。
1.1.3 邻硝基氨基氧化环化法
邻硝基氨基结构大量存在于含能化合物的结构当中,因此,邻硝基氨基氧化缩合法制备氧化呋咱结构具有非常广阔的应用前景,但迄今为止该方法在制备含氧化呋咱结构的含能材料中应用较为有限。次卤酸盐、碘代苯二乙酸酯(IBD)等均是邻硝基氨基缩合过程中常用的氧化剂,不同的氧化剂与氨基中孤对电子相互作用后通过环化形成氧化呋咱结构,但环化反应的转化机理存在差异:酸性条件下次卤酸盐为氧化剂时,氨基形成N-氧化结构,在硝基氧原子进攻下通过脱水反应形成氧化呋咱[25](Scheme 3a)。IBD 作为氧化剂的转化机理不同于次卤酸盐作为氧化剂的反应体系[26-27],Sako 等[28]以IBD 作 为 氧 化 剂,通 过 氢 化 锂(LiH)处理后的邻硝基氨基由IBD 完成氧化环化实现了氧化呋咱的构建(Scheme 3b)。此外,若以环糊精等作为添加剂,则可以促进脱水过程而在中性条件下完成该环化过程[29](Scheme 3c)。

Scheme 2 Oxidative condensation of 1,2-dioxime structure and the mechanism[20-24]

Scheme 3 Oxidative condensation of o-nitroamino structure and the mechanism[25-29]
1.2 基于1,3-偶极子分子间环加成方法
1,3-偶极子环加成反应(Huisgen 环加成反应)是制备五元杂环体系最高效的手段之一,具有反应清洁,转化率高等优点[30]。氧化腈的1,3-偶极子环加成机制是制备氧化呋咱结构最为有效的手段之一,其核心在于设计和选择何种前体来获得氧化腈的1,3-偶极子结构[31]。
1.2.1 氯肟碱性分子间缩合法
氯肟碱性缩合法机理是基于以氯肟转化所得氧化腈的1,3-偶极子环加成反应,即以两分子羟肟酰氯与弱碱(如碱金属碳酸盐)作用获取两分子氧化腈,这是一个原位反应获得1,3-偶极子过程,一分子氧化氰作为1,3-偶极子,另一分子氧化氰作为亲偶极体,通过氧化氰二聚反应完成环化获得氧化呋咱结构。在含能材料领域,制备氯肟最常用的策略是基于氰基的羟胺加成及氯代反应[32](Scheme 4)。氯肟碱性缩合法的突出优点在于前体制备方法成熟、反应条件极其温和、转化率高,因此底物适用性非常广泛。对称性是高能量密度材料设计的重要参数之一,与其他合成方法相比,氯肟碱性缩合法所得产物均为具有更高对称性的致密性结构,因而在对称型高能量密度材料的设计研发中具有重要的价值[33]。
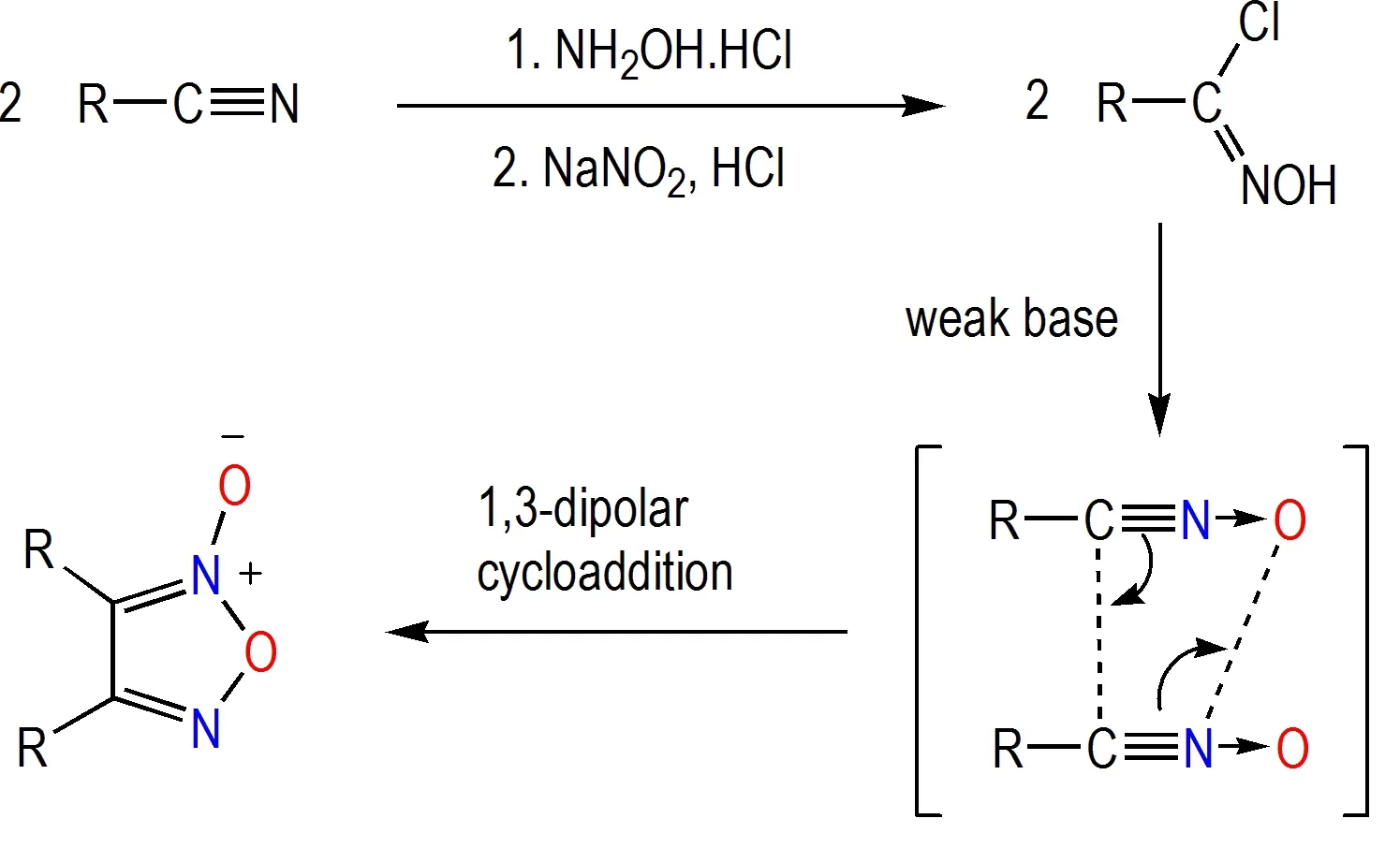
Scheme 4 Condensation of chloro-oxime under alkaline conditions and the mechanism[31-33]
与氯肟结构相似,硝基肟结构也是氧化腈的1,3-偶极子结构的有效前体。肟在四氧化二氮作用下可以转化为硝基肟结构,而硝基肟加热可以原位产生氧化腈的1,3-偶极子结构,进而发生1,3-偶极子环加成反应,获得氧化呋咱[34-35](Scheme 5)。
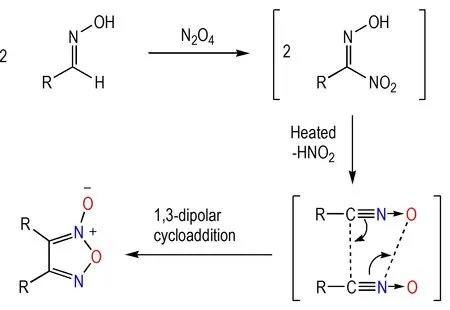
Scheme 5 Condensation through nitro-oxime and the mechanism[34-35]
1.2.2 分子间偕二硝甲基缩合法
偕二硝甲基缩合法是制备氧化呋咱的一类特殊方法,可通过脱除硝酸实现氧化腈的构建并基于其1,3-偶极子环加成反应获得氧化呋咱产物,可能的机理如Scheme 6 所示。相关研究表明当偕二硝甲基成盐后可通过四氧化二氮完成亚硝化,亚硝化产物连续2次脱除NO2自由基,形成氧化腈后进行1,3-偶极子环加成反应获得氧化呋咱产物。该缩合法适用于含有偕二硝甲基片段的化合物,并能够获得对称性的产物[36-37]。乙酸基缩合法可视为偕二硝甲基缩合法的衍生化策略:首先由α-位取代的乙酸在硝化条件下形成偕二硝甲基片段,进而通过与偕二硝甲基缩合法类似的过程形成氧化呋咱结构。典型的乙酸基缩合法包括氰基乙酸缩合制备二氰基氧化呋咱、叠氮基羰基乙酸缩合制备二叠氮基羰基氧化呋咱等,所得产物也均为对称性结构[38-39](Scheme 6)。
1.3 其它环化方法
烯烃加成转化法是基于烯烃与氮氧化物的加成反应,烯烃在亚硝化试剂作用下可以发生串联反应进而最终转化成为氧化呋咱结构。典型的一些转化包括:(a)以等当量吡啶的碱性条件下,四氟硼酸亚硝鎓与二取代苯乙烯反应形成相应氧化呋咱结构及其互变异构体结构[40-41]。(b)端烯在NaNO2/HOAc 作用下发生加成引入1,2-二亚硝基和2-硝基片段,其中1,2-二亚硝基异构化形成邻二肟结构环化为氧化呋咱[42-43]。此外,一些含肟类片段的特殊底物结构可以在特定条件下转化为氧化呋咱结构:(c)氯肟在二硝基甲烷碱金属盐的作用下形成类似的环化前体,经过环化形成硝基取代的氧化呋咱[44]。(d)以三乙烯二胺(DABCO)为碱,使用TsCl 对肟羟基中氧原子进行保护形成易离去的-OTs 基团,通过消除反应,可将邻硝基肟结构顺利转化为氧化呋咱结构[45](Scheme 7)。但需要指出,这些方法的应用范围具有一定局限性,因而尚未在氧化呋咱结构的制备中广泛应用。
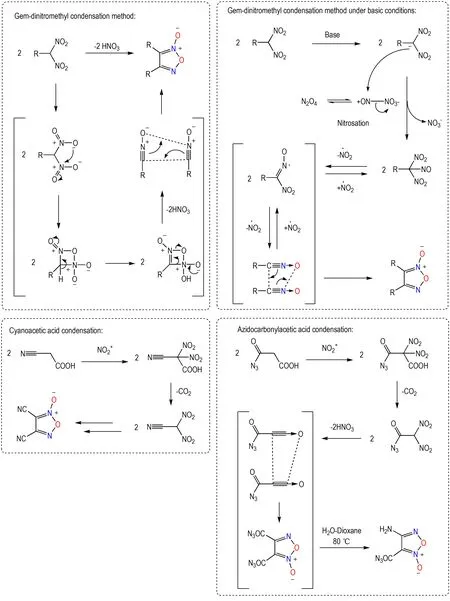
Scheme 6 Dinitromethyl condensation strategy and the mechanism[36-39]
2 典型氧化呋咱构建方法在含能材料合成中的应用
2.1 基于分子内环化法的氧化呋咱含能材料合成
具有富氮稠环骨架的含能材料普遍具有较低的冲击波感度和机械感度,以及更高的爆轰性能。邻硝基叠氮基脱氮缩合法是当前含氧化呋咱结构的富氮稠环含能材料合成中应用最多的方法。苯并三氧化呋咱(BTF)是重要的无氢炸药,能量密度水平和起爆性能突出,具有与奥克托今(HMX)相当的安全性、热安定性、爆轰特性,而其冲击起爆感度、熄爆直径与太安相当,作为起爆药被应用于导爆索装药和改善B 炸药的装药性能。经典的BTF 合成方法是基于酸性溶剂中三叠氮基三硝基苯加热至其熔点(131 ℃)的反应,该反应中相邻的硝基与叠氮基脱除氮气分子后形成环化产物[46-47]。为缩短BTF 合成步骤,避免使用感度较高的三叠氮基体系,Sheremetev 等[48]发展了离子液体中邻卤硝基苯直接一锅法合成苯并氧化呋咱的方法,并以此为基础实现了BTF 的新法合成(Scheme 8)。
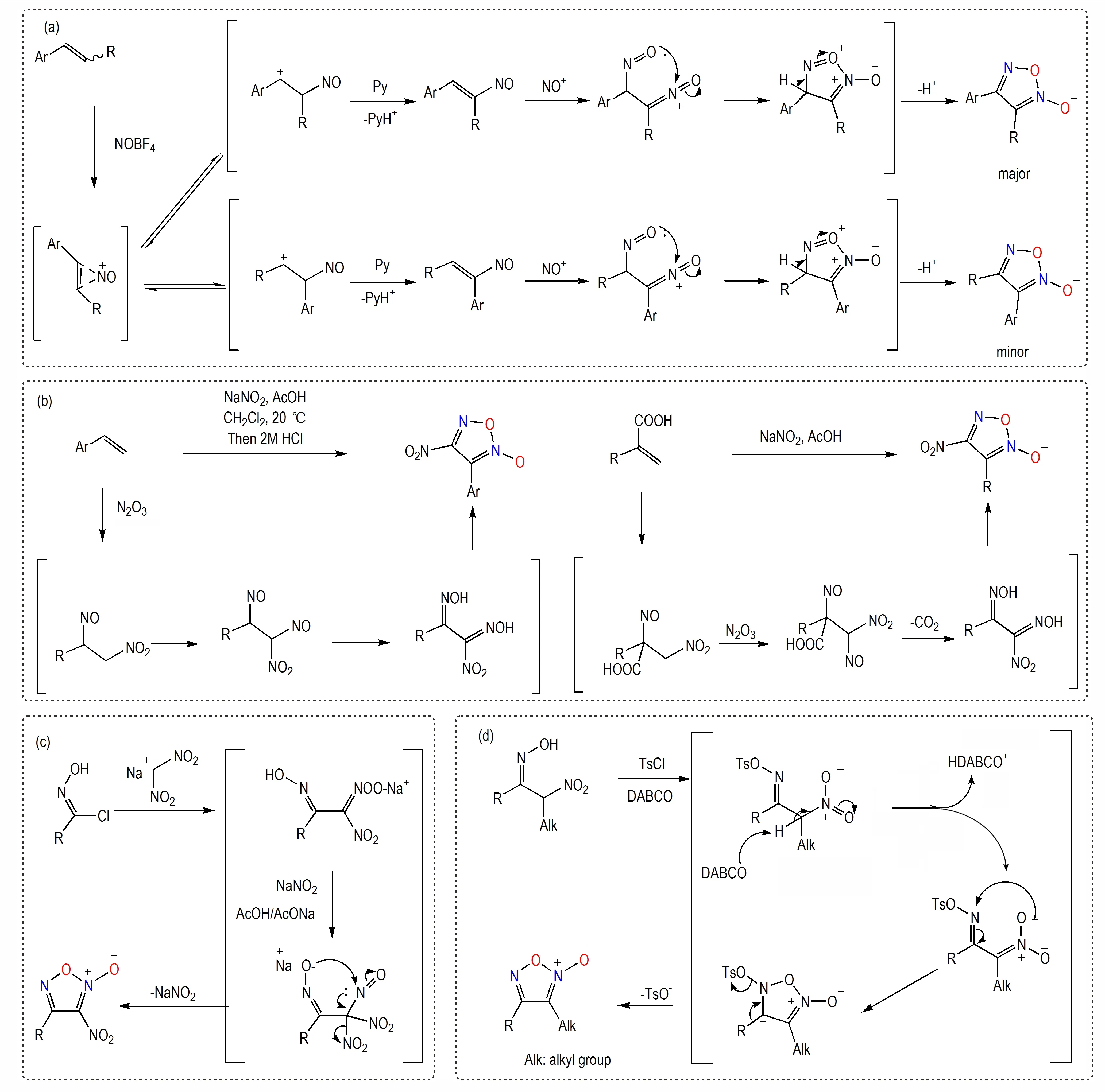
Scheme 7 Other conversion methods and mechanisms based on olefins, chloro-oximes and oximes[40-45]
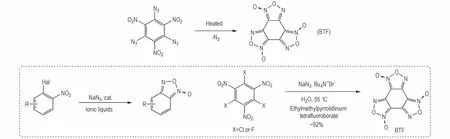
Scheme 8 Synthesis of furoxan and BTF by denitrogenation condensation of o-nitroazide structure[48]
氨基-硝基交替结构能够有效降低含能材料的机械感度,是设计钝感含能材料的重要途径。5,7-二氨基-4,6-二硝基苯并氧化呋咱(DADNB,CL-14)具有类似TATB 的氨基-硝基交替结构单元,作为重要的钝感炸药,其理论密度达到1.91 g·cm-3,理论爆速为8340 m·s-1[49-50]。CL-14 的合成是以三硝基氯苯为原料,利用叠氮化钠进行取代反应,形成邻硝基叠氮基结构,加热条件下脱除氮气,形成氧化呋咱。在此基础上,利用异常亲核取代反应(VNS)反应在苯环上引入了氨基结构[51]。霍欢等[52]利用1,3,5-三硝基-2,4-二氯苯为原料,经过叠氮化、脱氮环化合成6-硝基苯并二氧化噁二唑(呋咱)(NBDOF),通过NBDOF 进行VNS 反应在苯环引入氨基可得含能化合物7-氨基-6-硝基苯并氧化呋咱(ANBDF)(Scheme 9)。
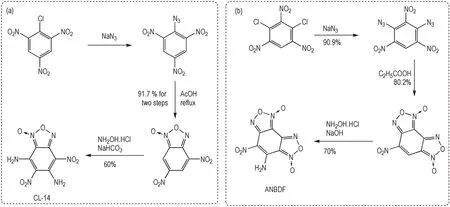
Scheme 9 Synthesis of CL-14 and ANBDF by denitrogenation condensation of o-nitroazide structure and VNS reaction[49-52]
总体而言,当前围绕氧化呋咱环系本身的构建方法研究以缩合反应和环加成反应为主,转化效率较高,且转化前体较为固定。发展更加多样的氧化呋咱环系构建方法目前仍是含能材料合成研究的重要方向。
Muralidharan 等[53]利用氯代 苯 并三唑底物进 行硝化、叠氮化形成邻硝基叠氮基结构,在加热条件下环化形成含氧化呋咱单元的富氮稠环结构。类似地,Churakov 等[54]利用二溴代的苯并二氧化四嗪底物进行硝化、叠氮化,在加热条件下环化形成氧化呋咱与二氧化四嗪一体的多N-氧化物富氮稠环结构。Korolev等[55]利用二溴代苯并咪唑底物进行硝化、叠氮化形成2 组邻硝基叠氮基结构,在加热条件下环化形成双氧化呋咱单元的富氮稠环结构,该双氧化呋咱单元可以发生互变异构形成混合物体系。Chavez 等[56]利用肼取代易离去的甲氧基,随后与亚硝酸反应在吡啶环中形成对称的2 组邻硝基叠氮基用于缩合反应,其中一组缩合形成了氧化呋咱单元,而另一组则缩合形成了四唑单元(Scheme 10)。
Klapötke 等[57]以二氯乙二肟为原料,在氰化钾作用下构建二氰基乙二肟,氰基部分在羟胺进攻下形成连续的2 组1,2-二肟单元,在溴氧化作用下形成4,4'-二氨基-3,3'-二氧化呋咱结构,对氨基氧化最终形成高能材料4,4'-二硝基-3,3'-二氧化呋咱(Scheme 11)。
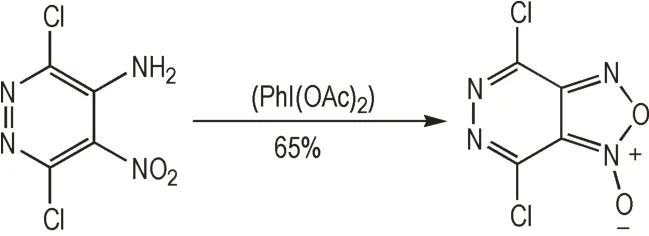
当前基于一般性缩合反应的方法的含氧化呋咱含能材料合成方法以双肟缩合法、邻硝基叠氮基缩合法为主,其他方法应用有限。这一方面源于相关前体制备的限制,另一方面是相关反应条件需要进一步根据底物结构进行筛选。例如,Rakitin 等[58]以4-氨基-3-硝基吡啶为原料,直接对邻氨基硝基片段进行氧化,形成了氧化呋咱片段。但该转化中氧化剂的选择至关重要,其中以(PhI(OAc)2)的氧化效率最高,但也仅达到65%(Scheme 12)。
2.2 基于1,3-偶极子环加成法在含能材料合成中的应用
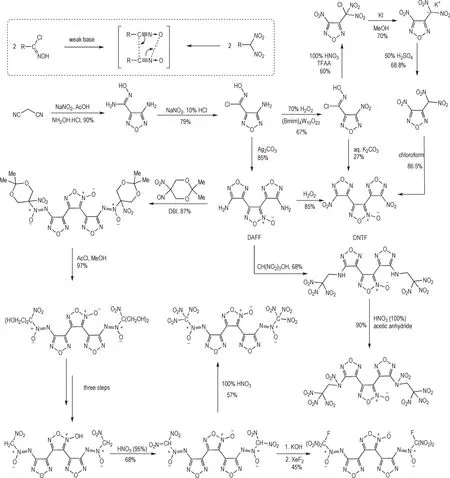
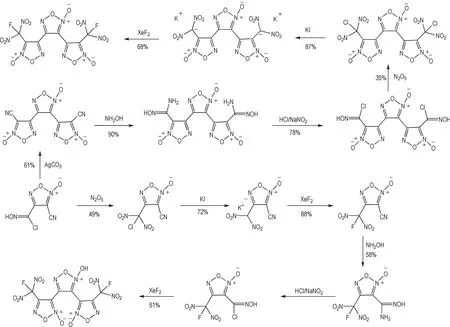
氯肟碱性缩合法是构建基于氧化呋咱结构的对称型含能材料最重要的途径,与其他方法相比,该方法的反应条件更加温和、转化率高,反应过程对其他含能结构的兼容性好,因而广泛的应用于对称型骨架的设计合成。氯肟碱性缩合法所获得的代表性骨架为基于碳碳单键所串联的呋咱-氧化呋咱-呋咱的三环结构,其中3,4-二硝基呋咱基氧化呋咱(DNTF)是典型的三代含能材料,具有较高的能量密度水平[59]。与基于氯肟缩合法构建DNTF 方法不同,Shreeve 等[60]以氨基氯肟呋咱结构为基础,探索了以偕二硝甲基缩合法构建DNTF 的可行性,即由氨基氯肟呋咱氧化所得的硝基氯肟呋咱中氯肟结构转化为偕二硝甲基结构,通过偕二硝基缩合将2 分子的硝基呋咱片段以氧化呋咱结构相连。而Luk'yanov 等[61-63]则以此类骨架为基础通过对致爆基团的改进获得了大量相似的具有较高能量密度水平的含能材料(Scheme 13)。而近年来,Shreeve等[64]及王伯周等[65]陆续发展了基于碳碳单键所串联的三氧化呋咱骨架,分别与硝基及氟偕二硝基型致爆基团结合,获得了一系列高性能含能材料体系,成为近年来含氧化呋咱体系含能材料研究的重要突破。与串联呋咱-氧化呋咱-呋咱骨架相比,串联的三氧化呋咱骨架其致密性和生成焓水平均显著提升[66],但由于多个氧化呋咱单元同时存在,整体稳定性有所降低且合成步骤较长,限制了此类串联的三氧化呋咱骨架含能材料的应用前景(Scheme 14)。
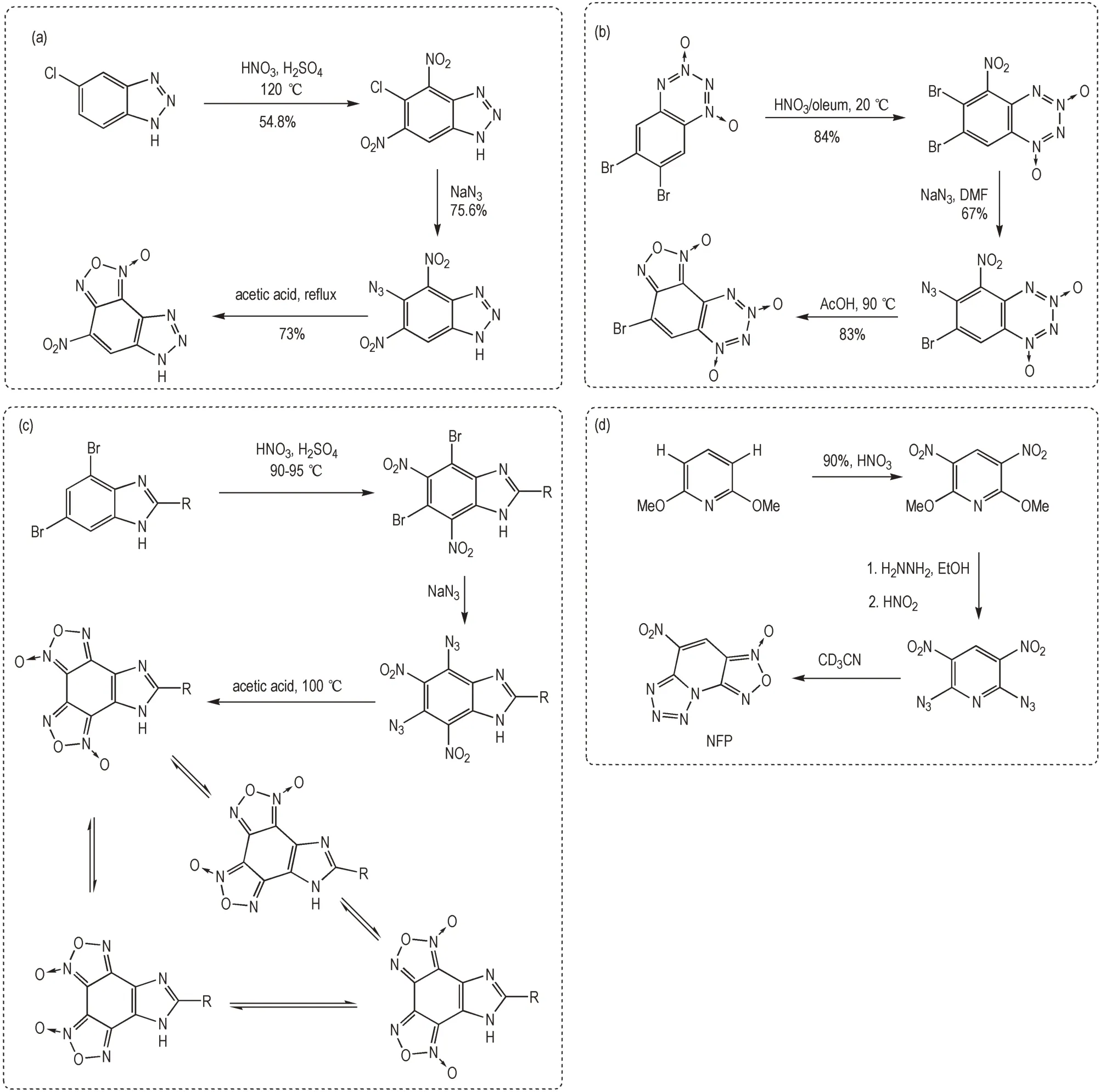
Scheme 10 Synthesis of nitrogen-rich fused aromatic energetic compounds by denitrogenation condensation of o-nitroazide structure[53-56]

Scheme 11 Synthesis of 4,4'-diamino-3,3'-bisfuroxan through oxidative condensation of 1,2-dioxime structure[57]
Scheme 12 4,7-Dichloro[1,2,5]oxadiazolo[3,4-d]pyridazine 1-oxide obtained by oxidation of o-nitroamino moiety[58]
Scheme 13 Tandem furazan-furoxan-furazan skeleton based energetic compounds obtained by different condensation reactions[59-63]
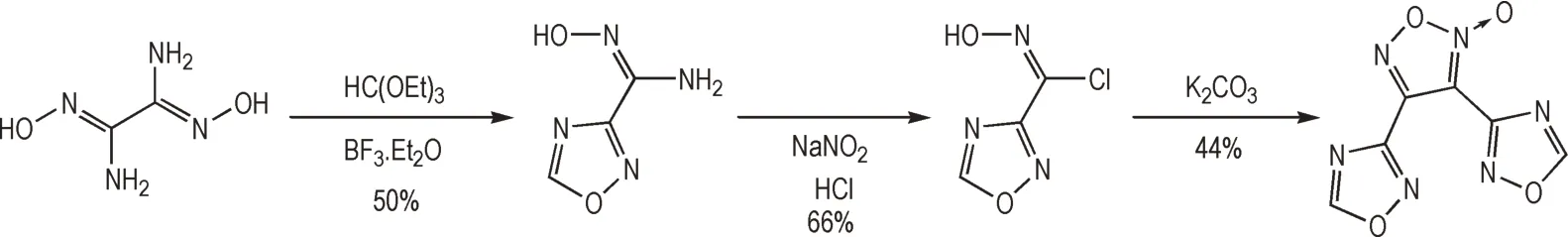
熔铸炸药是指熔化后可作为液相载体加入固相高能主炸药的一类炸药,其熔铸过程后期进一步再固化成型形成混合炸药,是军用混合炸药中极其重要的组分[67-69]。理想的载体炸药通常熔点低于110 ℃(由于通常以蒸汽进行熔化,因此最佳熔点范围为80~100 ℃)且要求熔点显著低于分解温度以避免在熔铸过程中发生分解。大量研究表明,以氧化呋咱结构为基础的含能材料往往具有较好的熔铸特性,因而广泛应用于军用混合炸药体系。DNTF 等以串联呋咱-氧化呋咱-呋咱为基础的多个含能化合物已成为被广泛关注的熔铸炸药,而串联三氧化呋咱体系同样表现出适宜的熔点水平,但分解温度偏低。基于异呋咱(1,2,4-噁二唑)的含能材料近年来被证明同样具有较低的熔点和较好的稳定性,因而由氧化呋咱与异呋咱环系串联所得的异呋咱-氧化呋咱-异呋咱结构成为熔铸炸药研究的另一重点潜在骨架[70]。围绕该骨架,Sabatini等[71-72]以二氨基乙二肟为原料,一单元的氨基肟结构与原甲酸三乙酯缩合形成异呋咱环结构,另一单元的氨基肟结构转化为氯肟结构后进行环加成反应形成氧化呋咱单元。该含能结构对冲击、摩擦和静电放电表现出优异的钝感特性,计算爆压比TNT 高20%,熔点114 ℃,被认为是理想的熔铸炸药组分(Scheme 15)。
Scheme 14 Tandem tri-furoxan skeleton energetic compounds obtained by condensation of chloro-oxime[66]
Scheme 15 Tandem Bis-(1,2,4-oxadiazolyl) furoxan skeleton energetic compounds obtained by condensation of chloro-oxime[71-72]
将氧化呋咱引入含能化合物体系对于提升整体能量密度水平具有重要意义。迄今为止,基于氧化呋咱的含能化合物种类仍较为有限,如何将氧化呋咱环系与其他富氮环系进行高效偶合以及如何将高性能致爆基团链接在氧化呋咱环上,应是下阶段该领域的重点内容。
3 结 论
氧化呋咱是重要的含能结构单元,具有高致密性、高氧平衡的特性,生成焓水平突出,是含能材料设计与研究的理想单元。本文总结了七类氧化呋咱的主要合成策略,涵盖了邻硝基叠氮基脱氮缩合法、1,2-二肟氧化缩合法、邻硝基氨基氧化缩合法、1,3-偶极子环加成法及其它合成方法,重点讨论了氧化呋咱构建的反应机理,并归纳了氧化呋咱典型构建方法在含能材料合成的研究进展。主要结论如下:
(1)邻硝基叠氮基芳香体系,反应条件温和,收率较高,是较为理想的氧化呋咱片段构建方法。但叠氮基硝基中间体感度普遍较高,危险性较大,而原位叠氮基取代并缩合过程可以避免了操作过程中的风险,具有更广阔的应用前景。
(2)双肟缩合、氯肟缩合、硝基肟缩合以及偕二硝基缩合等反应均属于1,3-偶极缩合反应过程,其反应机理类似,需根据具体的反应前体进行设计,但各自的底物适用性有一定差异。
(3)其它类型的氧化呋咱的制备方法具有一定的底物适用性限制,且反应条件具有各自的特点,虽然转化效率很高,但不属于通用型方法,需要依据具体的结构特征进行设计合成。
(4)氧化呋咱的热稳定性通常低于同类型的呋咱结构,在一定程度上影响了基于氧化呋咱骨架含能化合物的应用。
猜你喜欢
兵器装备工程学报(2022年12期)2023-01-06
化工管理(2021年7期)2021-05-13
商品与质量(2019年32期)2019-11-29
火工品(2019年3期)2019-09-02
成都信息工程大学学报(2019年5期)2019-05-21
火工品(2018年1期)2018-05-03
火炸药学报(2018年1期)2018-04-19
中国资源综合利用(2017年3期)2018-01-22
合成化学(2015年9期)2016-01-17
火炸药学报(2014年3期)2014-03-20
