
裂殖壶菌pks2基因启动子序列扩增及其活性分析❋
2023-02-21 03:15臧晓南王振东
中国海洋大学学报(自然科学版) 2023年1期
朱 清,臧晓南,王振东,马 帅
(中国海洋大学 海洋生物遗传学与育种教育部重点实验室,山东 青岛 266003)
启动子作为基因表达调控中必不可少的顺式作用元件,常用于基因重组表达等实验研究中,在构建表达载体时占据着重要的地位。启动子能够提供RNA聚合酶结合位点,控制着转录的起始[1-2]。在基因表达过程中,启动子的选择也发挥了很大的作用[3],选择启动活性高的强启动子一直是人们追求的目标。
染色体步移技术是在聚合酶链式反应(PCR)的技术基础上,从已知序列出发对邻近未知的序列进行探寻的方法,主要包括反向PCR,接头介导PCR以及半随机引物PCR等[4]。TAIL-PCR由Liu和Whittier[5]率先提出并成功应用于自动扩增和测序P1与YAC克隆细胞的插入端序列,以及拟南芥(Arabidopsisthaliana)T-DNA插入位点侧翼序列的分离和定位[6]。TAIL-PCR技术因其操作简便快捷,高效准确性高等优点在许多研究中被广泛应用。因此,本实验利用在半随机引物PCR基础上进行了改良的热不对称交错PCR技术(TAIL-PCR),使用3条方向相同且延伸温度较高的特异性引物与温度较低的兼并引物进行3次嵌套PCR反应,对基因未知序列进行扩增。
裂殖壶菌(Aurantiochytriumlimacinum)是一种海洋真菌,因其是DHA等多不饱和脂肪酸(PUFAs)高产菌而得到广泛关注。目前已有许多外源启动子运用于裂殖壶菌相关研究的实例,如郑楚强构建了裂殖壶菌中丙二酸单酰基转移酶(MAT)的过表达体系,利用TEF1启动子实现了MAT基因的过表达[7]。杨瑞雄等[8]同样利用外源TEF1启动子将希瓦氏菌属的Shewanellasp.SCRC2738的烯酰还原酶(ER)基因替换到裂殖壶菌(A.limacinum)SR21中异源表达,为调控裂殖壶菌中PUFAs的合成提供了新的思路。相较于外源启动子的广泛研究,对于裂殖壶菌内源启动子的探索目前还处于初步阶段,本实验室刘畅对裂殖壶菌pks1、pks3基因的启动子序列进行克隆重组,检测了启动子序列的活性,王振东在此基础上对两启动子序列进一步完善,得到了更多的顺式作用元件[9-10]。pks1、pks2和pks3是裂殖壶菌高效合成脂肪酸的PKS途径基因簇的3个基因,其中pks2基因的启动子序列目前尚未有研究。本研究拟对裂殖壶菌pks2基因的上游序列进行研究,探索裂殖壶菌的内源启动子的启动活性,为基因工程操作提供有效的基因元件。
1 材料与方法
1.1 实验材料
裂殖壶菌A.limacinumOUC168,大肠杆菌EscherichiacoliBL21及色氨酸缺陷型酵母SaccharomycescerevisiaeEBY100均为本实验室保存菌种;启动子检测载体pAcGFP1-1(见图1(a))以及genome walking试剂盒购自TAKARA公司,酵母表达载体pYD1(见图1(b))来自Invitrogen公司。克隆载体pEASY-Blunt-T simple Cloning Vector及菌株感受态E.coliTrans-T1来自于北京全式金公司。pks2基因为实验室前期克隆拼接获得[11]。实验中涉及到的引物列于表1。

(MCS:多克隆位点;AcGFP1:绿色荧光蛋白基因;SV40 poly(A)signal:SV40早期mRNA多腺苷酸化信号;f1 ori:f1单链DNA来源,包装AcGFP1-1的非编码链;AmpR promoter:氨苄西林抗性(β-内酰胺酶)启动子;SV40 ori:SV40复制起点;SV40 promoter:SV40早期启动子;NeoR/KanR:新霉素/卡那霉素耐药基因;HSV TK poly(A)signal:单纯疱疹病毒胸苷激酶多腺苷酸化信号;ori:pUC质粒复制起源。MCS: multiple cloning site; AcGFP1: Aequorea coerulescens green fluorescent protein gene; SV40 poly(A)signal: SV40 early mRNA polyadenylation signal; f1 ori: f1 single-strand DNA origin, packages noncoding strand of AcGFP1-1; AmpR promoter: Ampicillin resistance(β-lactamase)promoter; SV40 ori: SV40 origin of replication; SV40 promoter: SV40 early promoter; NeoR/KanR: Neomycin/Kanamycin resistance gene; HSV TK poly(A)signal: Herpes simplex virus thymidine kinase polyadenylation signal; ori: pUC plasmid replication origin.)
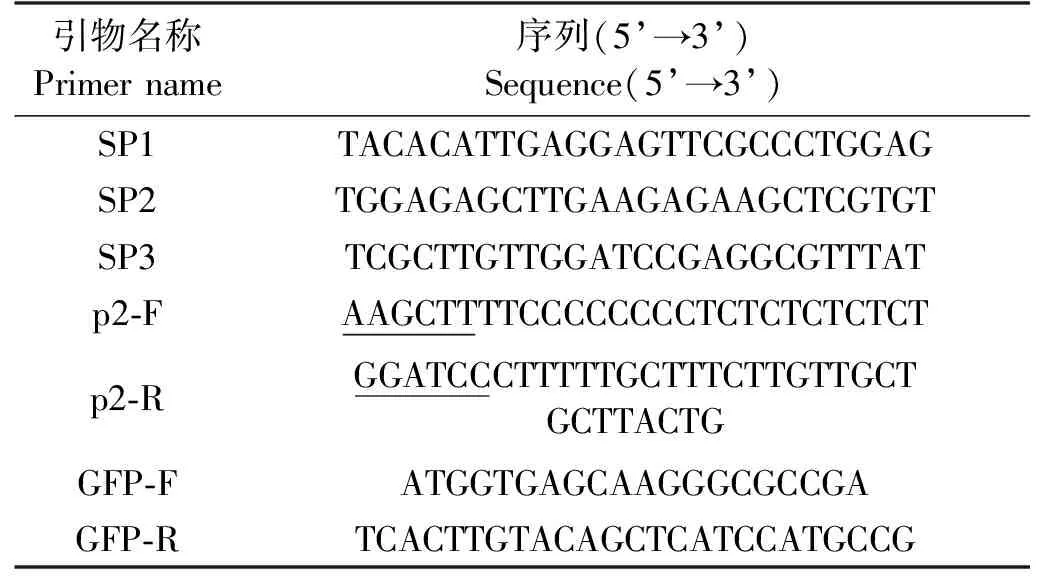
表1 引物列表
1.2 方法
1.2.1pks2基因5’端侧翼序列的获取与分析 根据pks2基因5’端序列设计3条退火要求温度较高且均为反向的特异性引物,分别命名为SP1、SP2与SP3(见表1),与genome walking试剂盒中的兼并引物进行3次巢氏PCR反应,得到目的侧翼序列。之后将序列通过PlantCARE[12]等进行分析预测,得到的pks2启动子序列命名为p2。
1.2.2pks2启动子的引物设计及连接克隆载体pEASY-Blunt-T simple Cloning Vector 根据得到的pks2启动子序列设计引物,上、下游分别加上BamHⅠ和HindⅢ酶切位点。上游引物为p2-F,下游引物为p2-R(列于表1,下划线为酶切位点序列)。以裂殖壶菌OUC168的DNA为模板扩得pks2启动子序列,并将其连接至pEASY-Blunt-T simpleCloning Vector并转入Trans-T1感受态细胞,测序准确的质粒命名为T-P2。
1.2.3pks2基因启动子连接pAcGFP1-1载体及转化大肠杆菌BL21 使用BamHⅠ和HindⅢ分别对T-P2和pAcGFP1-1载体进行双酶切,胶回收后利用T4连接酶连接,命名为pAcGFP-P2,转入制备好的E.coliBL21感受态细胞,筛选验证后将测序准确的菌株命名为E.coliBL21-pAcGFP-P2,同时将空载体pAcGFP1-1转化到E.coliBL21上,并将其命名为E.coliBL21-pAcGFP,用于后续对照。
1.2.4pks2基因启动子连接pYD1载体及转化S.cerevisiaeEBY100 使用NotⅠ和HindⅢ分别对pAcGFP-P2和pYD1载体双酶切,胶回收后连接测序,构建好的载体命名为pYD1-GFP-P2。利用酵母转化试剂盒将其转化到S.cerevisiaeEBY100上,经培养基平板筛选阳性转化子,之后提取转化子酵母基因组DNA,以其为模板进一步PCR验证测序,转化好的菌株命名为S.cerevisiaeEBY100-GFP-P2,同时使用NotⅠ和HindⅢ双酶切空载体pAcGFP1-1,得到绿色荧光蛋白gfp基因片段,将其连接pYD1载体再转入S.cerevisiaeEBY100用于后续对照,命名为S.cerevisiaeEBY100-GFP。
1.2.5 启动子的原核启动表达分析 将构建好的E.coliBL21-pAcGFP-P2和E.coliBL21-pAcGFP菌株与实验室前期保藏的含有pks1和pks3启动子的重组菌株(分别命名为E.coliBL21-pAcGFP-P1,E.coliBL21-pAcGFP-P3)一起接种至LB培养基中,37 ℃活化12 h,之后将活化好的菌液按1%的比例接种至新鲜培养基中继续培养5 h后测OD600值,将培养好的菌液经离心破碎后制备成蛋白上清样品,进行荧光分析以及SDS-PAGE和Western Blot检测。
1.2.6 启动子的真核启动表达分析 将构建好的S.cerevisiaeEBY100-GFP-P2,S.cerevisiaeEBY100-GFP菌株以及实验室前期构建好的含有pks1以及pks3启动子序列的重组酵母菌株(分别命名为S.cerevisiaeEBY100-GFP-P1,S.cerevisiaeEBY100-GFP-P3)一起进行表达。因pYD1质粒自身携带半乳糖诱导型启动子PGAL,而pks2为组成型启动子,所以为了避免PGAL启动子启动绿色荧光蛋白表达,重组菌株使用含2%葡萄糖的YNB-CAA培养基进行活化,30 ℃振荡培养24 h后,按10%比例进行转接,继续培养24 h后经离心破碎,取蛋白上清样品进行荧光分析,最后用SDS-PAGE和Western Blot检测。
2 结果与分析
2.1 pks2基因5’端侧翼序列的获取与分析
利用设计好的特异性引物与兼并引物进行3次PCR反应后,对第3次PCR产物进行琼脂糖凝胶电泳,电泳结果如图2。其中M泳道为marker,第1、2、3、4泳道分别为与试剂盒中4种兼并引物反应结果,除了第1泳道未出现条带外,其余3个泳道均出现了大小不一的条带,对这些条带进行胶回收后测序,通过与pks2基因5’端序列比对,最终确定得到了长度为409 bp的pks2基因5’端侧翼序列。

(M: Marker;1、2、3、4: 4种不同兼并引物PCR产物Reaction results of four different degenerate primers.)
将得到的pks2基因5’端侧翼序列运用PlantCARE进行预测分析(见图3),发现在序列中存在CAAT-box和TATA-box等常见的真核生物启动子作用元件,TATA-box具有确保转录准确开始的作用,而CAAT-box元件能有效的控制转录过程的速率[2,12]。此外,还发现有参与光响应的调节元件CAG-motif和G-box,以及MYB转录因子结合位点核心序列,厌氧诱导中重要的调节元件ARE等,初步推测这段序列可能具有启动活性,不仅是一个组成型启动子,还具备诱导型启动子的特点,在特定诱导条件下能够发挥功能。

图3 pks2基因启动子序列预测
2.2 原核表达载体构建及重组菌株转化
以裂殖壶菌OUC168的基因组DNA为模板,从pks2基因ATG前409 bp处开始扩得启动子序列。胶回收后连接克隆载体,进一步酶切后连接启动子检测载体pAcGFP1-1得到重组原核表达载体pAcGFP-P2,重组质粒图谱如图4所示。将pAcGFP-P2与空载体pAcGFP1-1分别转入提前制备好的E.coliBL21感受态细胞,对单一菌落转化子进行菌液PCR检测,检测结果如图5所示。图5(a)为载体pAcGFP-P2转E.coliBL21检测结果,使用引物P2-F和GFP-R分别对转化子和E.coliBL21进行PCR,除了泳道4E.coliBL21没有出现条带以外,其余3个泳道均出现了单一且清晰条带,且大小与pks2基因启动子和绿色荧光蛋白gfp基因两片段加起来的大小一致(1 129 bp),证明表达载体pAcGFP-P2成功转化到E.coliBL21中。图5(b)为使用绿色荧光蛋白gfp基因的上下游引物GFP-F与GFP-R对载体pAcGFP1-1转化检测结果,结果显示除泳道5以E.coliBL21为模板未出现条带外,泳道6、7均出现单一清晰条带,大小与gfp基因长度一致(720 bp),证明载体pAcGFP1-1也成功转化到E.coliBL21中,经进一步测序得到重组菌株E.coliBL21-pAcGFP-P2和E.coliBL21-pAcGFP。

图4 重组质粒pAcGFP-P2图谱

((a)载体pAcGFP-P2转化E. coli BL21菌PCR检测图。M:Marker;泳道1、2、3:pAcGFP-P2转化E. coliBL21阳性转化子;泳道4.:E. coli BL21菌株。(b)载体pAcGFP1-1转化E. coli BL21菌PCR检测图。M:Marker;泳道5:E. coliBL21菌株;泳道6、7:pAcGFP1-1转化E. coliBL21阳性转化子。(a)PCR result of E. coli BL21 transformed by vector pAcGFP-P2.M: Marker; Lanes 1, 2, 3: pAcGFP-P2 transformed E. coli BL21 positive transformant; Lane 4: E. coli BL21.(b)PCR result of E. coli BL21 transformed by vector pAcGFP1-1.M: Marker; Lane 5: E. coli BL21; Lanes 6, 7: pAcGFP1-1 transformed E. coli BL21 positive transformant.)
2.3 真核表达载体构建及酵母转化
对之前构建好的原核表达载体使用NotⅠ和HindⅢ双酶切,连接同样酶切后的pYD1载体,得到重组真核表达载体(见图6),之后进行酵母转化。以提取的转化子基因组DNA为模板进行PCR检测,检测结果如图7所示。图7(a)中使用引物P2-F和GFP-R,除第4泳道由于使用S.cerevisiaeEBY100的DNA为模板而未有条带外,其余3个泳道利用载体pYD1-GFP-P2转酵母的阳性转化子基因组DNA为模板均出现单一、明亮的条带,且大小与预期长度一致(pks2启动子和gfp基因共计约1 129 bp),证明载体pYD1-GFP-P2成功转化到S.cerevisiaeEBY100中,并插入其基因组中;图7(b)中使用引物GFP-F和GFP-R,除泳道5由于同样使用S.cerevisiaeEBY100为模板而未出现条带外,使用载体pYD1-GFP转化酵母的阳性转化子基因组DNA为模板的泳道6、7均出现了与绿色荧光蛋白gfp基因长度相符的单一条带(720 bp),证明载体pYD1-GFP也成功转入S.cerevisiaeEBY100中,通过进一步测序确定重组菌株S.cerevisiaeEBY100-GFP-P2和S.cerevisiaeEBY100-GFP。

图6 重组质粒pYD1-GFP-P2图谱

((a)载体pYD1-GFP-P2转化S. cerevisiae EBY100检测图。M:Marker;泳道1、2、3:pYD1-GFP-P2转化S. cerevisiae EBY100阳性转化子;泳道4:S. cerevisiae EBY100菌株。(b)载体pYD1-GFP转化S. cerevisiae EBY100检测图。M:marker;泳道5:S. cerevisiae EBY100菌株;泳道6、7:载体pYD1-GFP转化S. cerevisiae EBY100阳性转化子。(a)Detection diagram of vector pYD1-GFP-P2 transformed S. cerevisiae EBY100.M: Marker; Lanes 1,2,3: pYD1-GFP-P2 transformed S. cerevisiae EBY100 positive transformant; Lane 4: S. cerevisiae EBY100.(b)Detection diagram of vector pYD1-GFP transformed S. cerevisiae EBY100.M: Marker; Lane 5: S. cerevisiae EBY100; Lanes 6, 7: pYD1-GFP transformed S. cerevisiae EBY100 positive transformant.)
2.4 启动子的原核启动表达分析
2.4.1 重组菌株的荧光检测分析 对构建好的重组菌株E.coliBL21-pAcGFP-P2,E.coliBL21-pAcGFP以及实验室保存的E.coliBL21-pAcGFP-P1,E.coliBL21-pAcGFP-P3活化后进行荧光检测分析,结果如图8所示,在488 nm波长激发光下,菌株均呈现不同程度荧光发射峰,且在520 nm左右处出现最高峰,其中重组菌株E.coliBL21-pAcGFP-P2荧光活性最高,菌株E.coliBL21-pAcGFP-P3次之,然后为E.coliBL21-pAcGFP-P1菌株,而E.coliBL21-pAcGFP的荧光强度最低,表明pks1、pks2和pks3基因启动子都具有一定的启动活性,并不同程度启动了绿色荧光蛋白gfp基因的表达,其中pks2基因启动子活性最高,与其他结果间差异显著(P<0.01)。

(GFP:E. coli BL21-pAcGFP菌株;1-GFP:E. coli BL21-pAcGFP-P1菌株;2-GFP:E. coli BL21-pAcGFP-P2菌株;3-GFP:E. coli BL21-pAcGFP-P3菌株GFP: E. coli BL21-pAcGFP strain; 1-GFP: E. coli BL21-pAcGFP-P1 strain; 2-GFP: E. coli BL21-pAcGFP-P2 strain; 3-GFP: E. coli BL21-pAcGFP-P3 strain.)
2.4.2 重组菌株的SDS-PAGE分析以及Western Blot检测 对重组菌株经破碎离心后的蛋白上清样品制样后进行SDS-PAGE分析,结果如图9(a)所示,其中泳道1为E.coliBL21的总蛋白上清样品,泳道2、3、4分别为重组的E.coliBL21-pAcGFP-P1、E.coliBL21-pAcGFP-P2和E.coliBL21-pAcGFP-P3菌株总蛋白上清样品,与泳道1相比,其余3个泳道并无明显特异性条带出现,推测是由于表达量不高,因此SDS-PAGE条带不易辨认,进一步对样品使用绿色荧光蛋白gfp基因的特异性抗体进行Western Blot检测,结果如图9(b)所示,除泳道5E.coliBL21的总蛋白上清样品未出现明显条带外,其余3个泳道重组的E.coliBL21-pAcGFP-P1、E.coliBL21-pAcGFP-P2和E.coliBL21-pAcGFP-P3菌株总蛋白上清样品均在25~30 kDa之间出现一条明显的条带,与绿色荧光蛋白大小(26.7 kDa)相符,证明在原核菌株E.coliBL21中,pks1、pks2和pks3基因启动子均启动了绿色荧光蛋白gfp基因的表达。
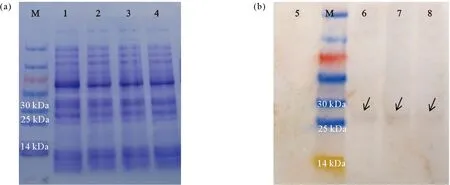
((a)原核重组菌株蛋白上清样品SDS-PAGE图。M:Marker;泳道1:E. coli BL21上清样品;泳道2:E. coli BL21-pAcGFP-P1上清样品;泳道3:E. coli BL21-pAcGFP-P2上清样品;泳道4:E. coli BL21-pAcGFP-P3上清样品。(b)原核重组菌株蛋白上清样品Western Blot图。M:Marker;泳道5:E. coli BL21上清样品;泳道6:E. coli BL21-pAcGFP-P1上清样品;泳道7:E. coli BL21-pAcGFP-P2上清样品;泳道8:E. coli BL21-pAcGFP-P3上清样品。(a)SDS-PAGE of prokaryotic recombinant strains protein supernatant sample.M: Marker; Lane 1: E. coli BL21supernatant sample; Lane 2: E. coli BL21-pAcGFP-P1 supernatant sample; Lane 3: E. coli BL21-pAcGFP-P2 supernatant sample; Lane 4: E. coli BL21-pAcGFP-P3supernatant sample.(b)Western Blot of prokaryotic recombinant strains protein supernatant sample.M: Marker; Lane 5: E. coli BL21 supernatant sample; Lane 6: E. coli BL21-pAcGFP-P1supernatant sample; Lane 7: E. coli BL21-pAcGFP-P2 supernatant sample; Lane 8: E. coli BL21-pAcGFP-P3supernatant sample.)
2.5 启动子系列重组菌株的真核表达分析
2.5.1 重组菌株的荧光检测分析 将构建好的酵母重组菌株S.cerevisiaeEBY100-GFP-P2和S.cerevisiaeEBY100-GFP以及之前保存的菌株S.cerevisiaeEBY100-GFP-P1和S.cerevisiaeEBY100-GFP-P3活化后表达制样,对得到的蛋白上清样进行荧光检测分析,结果如图10,在488 nm激发光下,菌株出现不同程度荧光发射峰,且在520 nm左右处出现最高峰。与对照菌株S.cerevisiaeEBY100-GFP相比,3种启动子均不同程度的启动了GFP的表达,其中菌株S.cerevisiaeEBY100-GFP-P2荧光略强,与其他结果间差异显著(P<0.05),证明pks2基因启动子活性最高,pks3基因启动子次之,pks1基因启动子活性最低,与在原核表达系统中荧光检测结果一致。
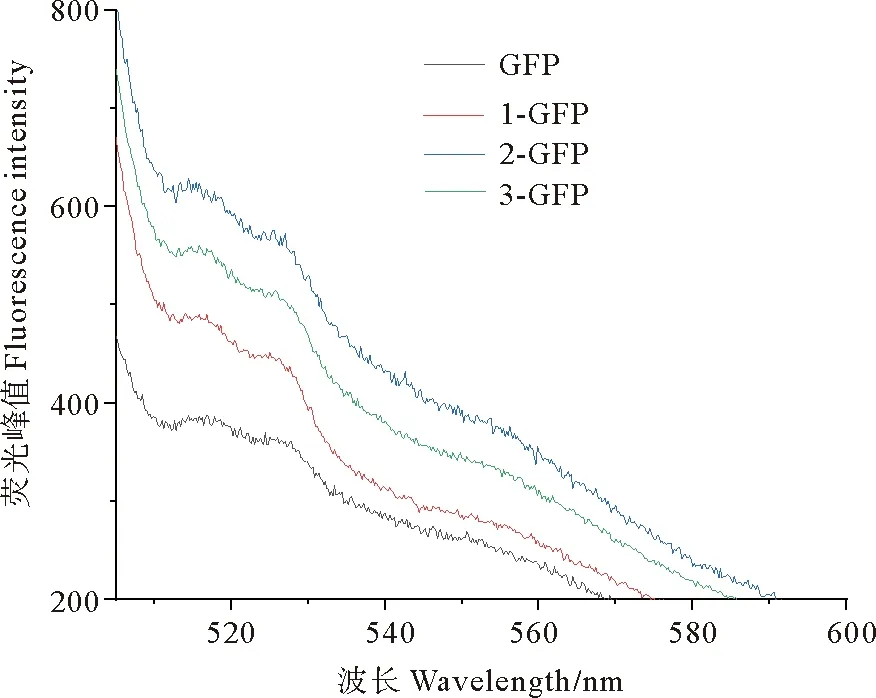
(GFP:S. cerevisiae EBY100-GFP菌株;1-GFP:S. cerevisiae EBY100-GFP-P1菌株;2-GFP:S. cerevisiae EBY100-GFP-P2菌株;3-GFP:S. cerevisiae EBY100-GFP-P3菌株。GFP: S. cerevisiae EBY100-GFP strain; 1-GFP: S. cerevisiae EBY100-GFP-P1 strain; 2-GFP: S. cerevisiae EBY100-GFP-P2 strain; 3-GFP: S. cerevisiae EBY100-GFP-P3 strain.)
2.5.2 重组菌株的SDS-PAGE分析以及Western Blot检测 将真核重组菌株蛋白上清制样后同样进行SDS-PAGE分析和Western Blot检测,结果如图11所示。图11(a)为真核重组菌株SDS-PAGE图,泳道1为S.cerevisiaeEBY100蛋白样,泳道2、3、4分别为重组菌株S.cerevisiaeEBY100-GFP-P1,S.cerevisiaeEBY100-GFP-P2,S.cerevisiaeEBY100-GFP-P3的蛋白上清样。SDS-PAGE结果显示,在25~30 kDa之间,条带较为紧密,重组菌株无法判断出有明显区别于S.cerevisiaeEBY100的条带出现。图11(b)为利用绿色荧光蛋白gfp基因特异性抗体进一步进行的Western Blot检测,与S.cerevisiaeEBY100菌株上清样(泳道5)相比,重组菌株S.cerevisiaeEBY100-GFP-P1(泳道6)、S.cerevisiaeEBY100-GFP-P2(泳道7)和S.cerevisiaeEBY100-GFP-P3(泳道8)均出现条带,且大小与绿色荧光蛋白大小一致(26.7 kDa),证明pks1、pks2和pks3基因启动子都启动了gfp基因的表达,具有启动活性。
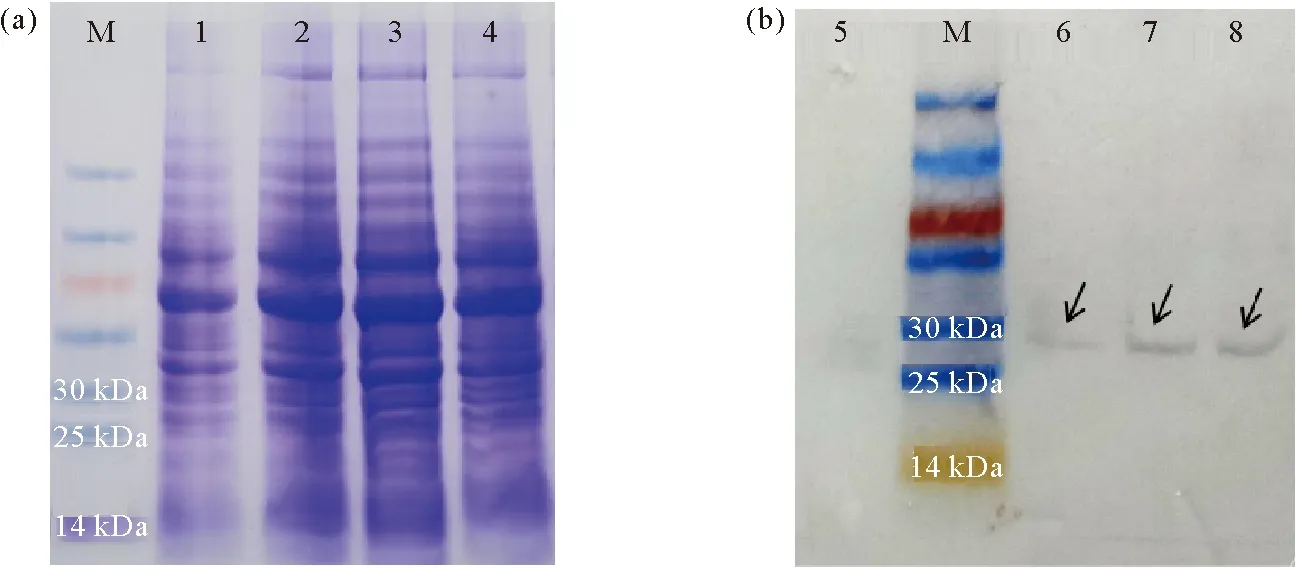
((a)真核重组菌株蛋白上清样品SDS-PAGE图。M:Marker;泳道1:S. cerevisiae EBY100上清样品;泳道2:S. cerevisiae EBY100-GFP-P1上清样品;泳道3:S. cerevisiae EBY100-GFP-P2上清样品;泳道4:S. cerevisiae EBY100-GFP-P3上清样品。(b)真核重组菌株蛋白上清样品Western Blot图。M:Marker;泳道5:S. cerevisiae EBY100上清样品;泳道6:S. cerevisiae EBY100-GFP-P1上清样品;泳道7:S. cerevisiae EBY100-GFP-P2上清样品;泳道8:S. cerevisiae EBY100-GFP-P3上清样品。(a)SDS-PAGE of eukaryotic recombinant strains protein supernatant sample.M: marker; Lane 1: S. cerevisiae EBY100 supernatant sample; Lane 2: S. cerevisiae EBY100-GFP-P1 supernatant sample; Lane 3: S. cerevisiae EBY100-GFP-P2 supernatant sample; Lane 4: S. cerevisiae EBY100-GFP-P3 supernatant sample.(b)Western Blot of prokaryotic recombinant strains protein supernatant sample.M: Marker; Lane 5: S. cerevisiae EBY100 supernatant sample; Lane 6: S. cerevisiae EBY100-GFP-P1 supernatant sample; Lane 7: S. cerevisiae EBY100-GFP-P2 supernatant sample; Lane 8: S. cerevisiae EBY100-GFP-P3 supernatant sample.)
3 讨论
本研究利用TAIL-PCR技术,通过3次巢氏PCR反应,扩增出了pks2基因5’端侧翼序列,经分析初步确定了这段序列具有启动子活性。在对pks2基因启动子序列活性的进一步研究中,选用了绿色荧光蛋白gfp基因,为探索在不同宿主中启动子的活性提供了便利条件;同时通过选择常用的原核大肠杆菌表达系统以及真核酿酒酵母表达系统,发现在E.coliBL21和S.cerevisiaeEBY100中,pks2基因启动子均不同程度的启动了绿色荧光蛋白gfp基因的表达,这说明pks2在原核表达系统和真核表达系统中都具有启动活性。
裂殖壶菌具有高效合成脂肪酸的聚酮合酶途径(PKS途径),pks1、pks2和pks3组成了PKS途径相关酶的基因簇。目前已有许多针对参与PKS途径关键基因和结构酶域的研究,揭示了参与PKS途径的基因的具体功能以及改造可能性,为构建高产DHA的优势菌株奠定了理论基础。本实验室刘畅[9]克隆了裂殖壶菌pks1、pks3基因的启动子序列,王振东[10]在此基础上对两启动子序列进一步延长,发现这两个启动子的启动活性并不高,而pks2基因的启动子目前还没有被研究。在对裂殖壶菌进行基因改造获得高产DHA菌株的研究中,常常存在外源基因表达不够理想的情况,选择一个合适的强启动子,往往能够提高重组蛋白的表达[13]。Han等[14]通过建立β-半乳糖苷酶报告系统,比较了在裂殖壶菌中4种启动子的启动活性,利用活性最强的ccg1p启动子,实现了裂殖壶菌中ATP-柠檬酸裂解酶和乙酰辅酶a羧化酶的过量表达。除了外源启动子的筛选,选用比外源启动子活性更高的内源性启动子往往能达到事半功倍的效果。李杰等[15]在对转基因盐藻外源基因表达的研究中发现,利用盐藻的诱导型内源启动子比外源启动子更有利于外源基因的表达;谭玮[16]在对蓝藻中外源基因人粒-巨噬细胞集落刺激因子的表达研究中,使用了蓝藻的2种内源启动子,与使用外源启动子相比,均在一定程度上提高了基因的表达。江贤章等[17]通过对海洋破囊壶菌Thraustochytriumsp.FJN-10的Δ4-脂肪酸脱饱和酶5’端侧翼序列扩增分析,得到具有启动子活性的序列;夏晓峰[18]又针对破囊壶菌Thraustochytriumsp.FJN-10的Δ5-脂肪酸脱饱和酶5’端侧翼序列进行扩增,同样得到了一段具有启动子活性的序列,为从基因水平研究破囊壶菌Thraustochytrium中DHA的生物合成奠定了基础。实验中扩得的pks2基因启动子,作为裂殖壶菌的内源启动子或许可以用于后续的研究。
在对启动子活性的进一步比较分析中,发现新得到的pks2基因启动子具有比pks1、pks3基因启动子更高的启动活性。经过序列比对分析发现pks1、pks2和pks3基因的启动子序列中都具有CAAT-box,但是CAAT-box的位置有所不同。其中pks2启动子序列中的CAAT-box距离转录起始点的距离更近,因而这可能使其调控效率更高。此外pks2启动子序列中还有参与光响应的调控元件CAG-motif、G-box等,这可能使转录更容易受到调控,从而具有更高的转录效率。
新扩增得到的pks2基因启动子虽然包含的顺式作用元件并不多,但仍显现出较高的启动活性,在未来对其进一步研究或许存在更完整序列与更高活性。总体来说,作为裂殖壶菌以外其他宿主的外源启动子,甚至作为裂殖壶菌内源启动子,pks2基因启动子的获得与成功表达显示出其在启动基因表达中具备的潜力,为裂殖壶菌代谢调控和合成生物学研究提供了基因资源。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
华人时刊(2022年9期)2022-09-06
华人时刊(2020年15期)2020-12-14
数学大王·低年级(2020年8期)2020-08-14
生物工程学报(2019年1期)2019-01-30
江西医药(2018年8期)2018-10-24
罕少疾病杂志(2017年2期)2017-02-23
小雪花·初中高分作文(2016年9期)2016-05-14
中国火炬(2013年11期)2013-07-25
