
22例儿童急性痘疮样苔藓样糠疹临床与病理分析
2023-02-20 06:19钱莹莹李纪兵吴亚芬
中国麻风皮肤病杂志 2023年3期
钱莹莹 钱 华 李 巍 鲁 慧 胡 翠 张 婷 李纪兵 吴亚芬
苏州大学附属儿童医院,江苏苏州,215000
急性痘疮样苔藓样糠疹(pityriasis lichenoides et varioliformis acuta, PLEVA)是一种病因不明、临床相对少见的炎症性皮肤病。PLEVA起病急,病程长短不一,常为鳞屑性红斑、丘疹、丘疱疹、水疱、坏死、结痂等多形性损害。PLEVA虽少见但在儿童中并不罕见,现对2013年7月至2022年7月至我科就诊的22例儿童PLEVA患者的临床资料及组织病理进行系统分析,总结儿童PLEVA的临床及组织病理特点。
1 临床资料
1.1 一般资料 收集2013年7月至2022年7月至苏州大学附属儿童医院皮肤科就诊并经组织病理确诊的22例PLEVA患者。22例患儿中男16例(72.73%),女6例(27.27%),男女比例为2.67∶1。发病年龄4~13岁,平均年龄(8.77±2.84)岁,其中4~5岁5例(22.73%),9~10岁7例(31.82%)。病程1周~12个月。
诊断标准[1,2]:(1)急性发病,皮损广泛且多形性表现,常表现为鳞屑性红斑、丘疹、丘疱疹,伴不同程度的坏死结痂,皮损陆续成批出现,新老皮损可同时存在。(2)组织病理特征:角化不全,表皮水肿,淋巴细胞、红细胞游入表皮,基底细胞液化变性,真皮浅层水肿,毛细血管扩张,血管周围淋巴细胞浸润。
1.2 临床表现 儿童PLEVA典型的皮损常为多形性损害,表现为泛发的米粒至黄豆大鳞屑性红斑、丘疹、丘疱疹,伴不同程度的坏死结痂(图1)。按照Gelmetti等[3]制定的分型标准:中心型:累及面部、躯干和腹股沟;弥漫型:躯干和四肢均受累;外周型:四肢及掌跖受累。22例患儿中18例(81.82%)为弥漫型,3例(13.64%)为中心型,1例(4.55%)为外周型。根据皮损数目分为3级:0~50个皮损为1级,51~100个为2级,>100个为3级。22例患儿中,10例患儿皮损密集分布>100个为3级(A组),8例2级(B组),4例1级(C组)。发疹前16例(72.73%)患儿有感染史,其中12例有发热、咽痛或上呼吸道感染病史,1例有EB病毒感染病史,1例有手足口病病史,2例有流行性腮腺炎病史。
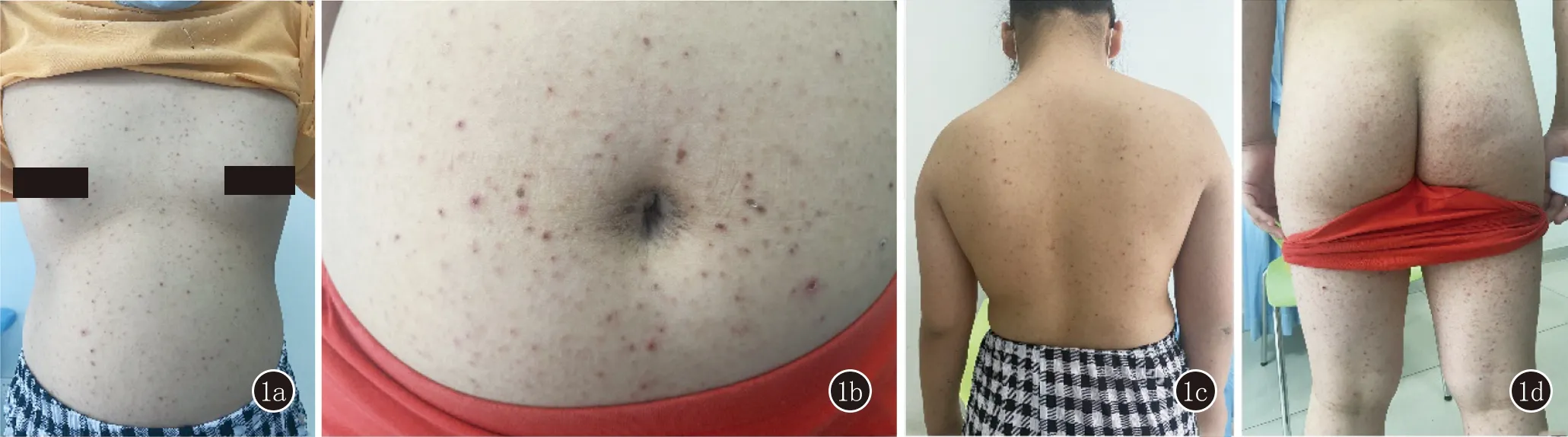
图1 躯干、四肢泛发米粒至黄豆大鳞屑性红斑、丘疹、丘疱疹,伴不同程度的坏死、结痂
1.3 实验室检查 22例患儿均行血尿常规、肝肾功能、血沉及抗O检查,18例血常规显示白细胞升高(11×109/L~18.17×109/L),同时伴有中性粒细胞或淋巴细胞计数及百分比升高,1例出现血小板减少(78×109/L),4例AST(60~152 U/L)、ALT(48~178 U/L)升高,3例抗O升高(368~560 IU/mL),8例血沉增快(30~80 mm/h)。10例患儿行腹部彩色超声检查,1例患儿有肝脾肿大。
1.4 组织病理 22例患儿均行皮肤病理活检,因皮损多样性,病程中皮损演变,临床取材,其组织学表现差异较大。皮损组织病理主要表现为表皮角化不全,表皮细胞间及细胞内水肿,可见散在坏死的角质形成细胞,基底细胞不同程度的液化变性,真皮内淋巴细胞呈楔形浸润,可见真皮乳头层红细胞外溢,部分红细胞移入表皮(图2)。
1.5 临床误诊情况 22例患儿中,14例(63.64%)患儿首诊临床误诊,其中误诊为点滴状副银屑病2例,水痘3例,丘疹性荨麻疹3例,玫瑰糠疹3例,淋巴瘤样丘疹病1例,血管炎2例。
1.6 治疗情况 22例患儿均采用联合治疗,根据其皮损严重程度、年龄、是否合并有系统症状制定个体化治疗方案,主要药物为红霉素30~50 mg/(kg·d)分3次口服,复方甘草酸苷注射液1~2 mL/(kg·d)静滴,部分皮损泛发、病情较重者予以甲泼尼龙1 mg/(kg·d),静脉丙种免疫球蛋白400 mg/(kg·d)静滴,所有患儿均应用抗组胺药、外用药物及其他对症处理。疗效判定标准:临床症状消失,皮损消退>80%为痊愈;临床症状部分消退,皮损消退面积50%~80%为有效;临床症状改善不明显,皮损消退面积<50%为无效。见表1。

2a~2c:角化过度伴角化不全,颗粒层减少,棘层轻度肥厚伴棘细胞间轻度水肿,基底细胞液化变性,真皮乳头轻度水肿伴有红细胞外渗,真皮血管及附属器周围以淋巴细胞为主的炎细胞浸润(2a:HE,×40;2b、2c:HE,×100);2d:真皮血管及附属器周围可见淋巴细胞浸润(HE,×200)

表1 22例患儿治疗情况及疗效 例
1.7 随访 22例患者随访时间6个月~2年,20例患儿随访期间无复发,1例患儿在停药2个月之后躯干部出现少量皮损,后口服少剂量糖皮质激素,皮损消退,缓慢减量后停药。1例患儿在停药8个月后复发,口服复方甘草酸苷片、红霉素,辅以外用药以后皮损消退。该2例患儿后均随访无复发。
2 讨论
PLEVA又称为Mucha-Habermann病,属于苔藓样糠疹的一型。除了亚型发热性溃烂坏死性Mucha-habermann(febrile ulcerative mucha-haberman disease, FUMHD)病情严重,可至死亡,PLEVA一般预后较好,具有一定的自限性[4]。PLEVA可在任何年龄发病,男性发病多,好发于20~30岁的青壮年,但在儿童中PLEVA并不罕见。Zang等报道对75例苔藓样糠疹的长期随访研究中,65例患者为儿童[5]。本研究中的22例患儿,男16例,女6例,男女比例2.67∶1,这与既往文献报道相似[6]。平均年龄(8.77±2.84)岁,其中4~5岁5例(22.73%),9~10岁 7例(31.82%),提示4~5岁和9~10岁是发病的高峰年龄,既往研究中发病的高峰年龄也不尽相同,徐秀莲等[7]报道4~5岁和12~13岁为发病的高峰阶段,而Ersoy-Evans[8]等发现2~3岁和5~7岁为发病高峰。
PLEVA目前发病机制不明,主要有感染、T细胞异常增殖、免疫复合物介导的超敏反应三种学说。可能是由感染相关因子,触发了继发于T细胞的炎症反应,由免疫复合物介导的超敏反应性血管炎。目前已发现的与PLEVA发病相关的病原体有水痘带状疱疹病毒、单纯疱疹病毒、EB病毒、溶血性链球菌、细小病毒等[9],本研究中有16例(72.3%)患儿发病前有感染病史,12例为上呼吸道感染史,1例有EB病毒感染,1例有手足口病病史,2例有流行性腮腺炎病史,说明感染为儿童PLEVA发病的重要诱发因素。实验室检查中发现18例患儿血常规异常,提示有细菌或病毒感染,3例抗O增高提示发病前有链球菌感染,1例患儿出现血小板减少及4例患儿出现转氨酶升高,均为病情相对较重及皮损泛发者,1例发现脾脏肿大的患儿为发病前EB病毒感染史。典型的PLEVA皮损表现为淡红色或者红褐色米粒至豌豆大的斑疹、丘疹、丘疱疹、水疱,中央可见出血,坏死,黑色结痂。常累及躯干及四肢近端,黏膜、掌跖及面部较少累及。本研究18例(72.73%)患儿皮损为弥漫型,说明儿童PLEVA的皮损常泛发,因PLEVA皮损的多形性,早期可模仿多种其他疾病,因此临床误诊率高,本研究中首诊误诊率高达63.64%。皮肤病理为诊断金标准,但因病理取材的偏差,临床表现与组织病理充分联系是确诊该病的必要条件。本研究的22例患儿均行皮肤病理活检,PLEVA的组织病理主要表现为:表皮角化不全,表皮细胞间及细胞内水肿,可见散在坏死的角质形成细胞,基底细胞不同程度的液化变性,真皮内淋巴细胞呈楔形浸润,可见真皮乳头层红细胞外溢,部分红细胞移入表皮。其中基底细胞的液化变性,真皮内血管周围炎是诊断PLEVA的必要条件。
目前PLEVA尚无统一的治疗方案,目前推荐的一线诊疗方案有紫外线疗法如NB-UVB或者PUVA,口服四环素或红霉素联合外用糖皮质激素可以获得较为满意的结果,另外也可使用免疫抑制剂甲氨蝶呤、环孢素等治疗[10]。在本研究中,考虑到儿童用药的安全性及治疗的配合性,并根据患儿皮损的严重程度,病情的轻重选择不同的联合治疗方案。主要治疗方案为红霉素30~50 mg/(kg·d)口服,复方甘草酸苷注射液 1~2 mL/(kg·d)静滴。部分皮损泛发,病情较重者加甲泼尼龙1 mg/(kg·d),静脉丙种球蛋白400 mg/(kg·d)静滴。20例(90.91%)治愈,说明儿童PLEVA预后好。22例患儿均完成半年以上的随访,有2例患儿分别在完全停药2个月和8个月后复发,后门诊口服药物后无复发。有文献报道PLEVA病情较长迁延,随访中发现有演变为蕈样肉芽肿或者淋巴瘤[11],因此PLEVA的患儿需要更为长期的随访。另外我们发现皮损愈合后,痘疮样的瘢痕及色素减退很难消除,损容性的外观影响儿童社交及心理健康。
因此PLEVA虽具有自限性,但在儿童患者中,PLEVA通常皮损泛发,早期行皮肤病理活检,尽早干预治疗对疾病预后较为关键,感染为儿童PLEVA的主要诱发因素,红霉素可作为儿童PLEVA中抗感染的首选药物,安全有效,另外PLEVA病程较长,可能演变、复发,患儿需要长期随访。
猜你喜欢
建材发展导向(2021年14期)2021-08-23
现代临床医学(2021年4期)2021-07-31
中国生殖健康(2020年2期)2021-01-18
祝您健康·文摘版(2019年4期)2019-06-11
中国生殖健康(2018年2期)2018-11-06
西南国防医药(2016年7期)2016-12-01
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
中国房地产业(2016年9期)2016-03-01
实用手外科杂志(2015年3期)2015-08-27
西部皮革(2015年3期)2015-04-16
