
小头骨发育不良原始侏儒症Ⅰ型的诊断(附1例报告)
2023-02-18 10:40:06王运程云张帆杨武
山东医药 2023年4期
王运,程云,张帆,杨武
1 蚌埠医学院研究生院,安徽蚌埠 233030;2 安徽医科大学附属六安医院儿科
小头畸形骨发育不良原始侏儒症I 型(MOPDⅠ)或Taybi-Linder综合征是一种罕见的常染色体隐性遗传性骨骼发育不良疾病,最早于1967 年被描述。患者表现为严重的宫内和出生后发育迟缓、小头畸形、面部畸形(前额倾斜、眼睛突出和小颌畸形)、稀疏的头发和干燥的皮肤[1]。放射学表现包括伴有椎弓裂的骨骼发育不良、水平髋臼、短而弯曲的长骨[2]。神经学表现通常包括严重的智力发育迟缓、白内障、听力障碍、中枢神经系统畸形、早发性癫痫和神经内分泌功能障碍[3]。根据人类基因突变数据库,目前约有12 种不同的RNU4ATAC 基因突变和50 例MOPD1 患者被报道。该病病情通常较严重,患者一般不会存活到3岁以上。目前,已报告了部分临床表型较轻的病例[4]。本病国内罕见,现报道1 例RNU4ATAC 基因突变导致婴幼儿小头畸形骨发育不良原始侏儒症I 型(MOPD1),以期为该病的临床诊治提供参考。
1 资料与方法
1.1 临床资料 先证者为男童,1 月27 天,系“发热、咳嗽2 天,呻吟半天”于安徽医科大学附属六安医院就诊,病程中有反复发热及咳嗽,入院前患儿开始出现呻吟,拒食,精神欠佳,时有震颤,拟“婴儿支气管肺炎;小头畸形”收住儿科。追问病史,患儿母亲孕检结果:NT:3.3 mm,唐筛18-三体临界风险,羊水穿刺提示样本的染色体在检测范围内未见异常,四维彩超提示胎儿颅骨内结构异常(BLAKE 囊肿待排,胼胝体缺失可疑,左侧颅内囊实性回声,鼻骨发育不良,SD: 4. 1-4. 5),胎盘血窦,胎儿肠管内回声增强,后复查四维彩超及晚期多次产科彩超均提示结构异常,因孕母不良孕产史(第一胎小头畸形夭折),妊娠期高血压、癫痫、胎儿生长受限、羊水过少。足月顺产,出生APgar 评分1、5、10 min 分别为8、10、10 分,出生体质量:1 940 g,小于同胎龄儿出生体质量的第10 百分位,头围小(29 cm),小颌畸形,耳位低平,皮肤干燥,毛发稀疏,羊水清,脐带无异常,胎盘血窦,原始反射引出不全。生后不久即出现呻吟症状,当时完善头颅CT 示脑实质体积缩小,脑沟裂增深、增宽,双侧脑室分离,幕上、下脑室系统扩张,中线结构尚居中。
1.2 入院查体及辅助检查 入院查体示神清,精神萎靡,呼吸稍促,发育落后,小头畸形,头围31 cm,小颌畸形,耳位低平,皮肤干燥,毛发稀疏,全身弥漫性红色皮疹,前囟近闭,巩膜黄染,双眼球突起,左侧明显,双瞳孔等大等圆,光反射存在,双上肢搐搦,肌张力增高明显,双手握拳,手指短细,双下肢肌力肌张力尚可,左下肢散在瘀斑,NS(-)。无通贯手,拥抱、吸吮、握持及觅食等原始反射可引出,双侧病理反射阴性。完善电解质检查,结果示电解质紊乱,凝血功能检查示APTT 及TT 极高,血气分析大致正常范围内;血常规检查示白细胞升高,中性粒细胞为主;肝功能检查示转氨酶及胆红素增高,甲功七项结果正常。
1.3 诊疗经过 入院后予中心吸氧改善症状,心电监护监测生命体征变化,头孢哌酮舒巴坦钠抗感染、维生素C营养心肌、谷胱甘肽保肝、葡萄糖酸钙纠正低钙血症、补液维持水电解质平衡、雾化对症治疗,3 d后患儿热退,咳嗽好转,家长要求出院,予以办理。
1.4 基因检测 经患儿家长知情同意后,分别留取患儿及父母EDTA抗凝血各2 mL,标本送至上海韦翰斯生物医药科技有限公司,对受检者家系临床全外显子组及毗邻剪接区域进行基因变异分析,发现受检者携带编码小核RNA(snRNA)基因 RNU4ATAC上的2个杂合变异,即 M1:n.51G>A 和 M2:n.55G>A。测序结果显示这两个变异分别遗传自母亲和父亲,构成复合杂合变异(家系图、一代测序峰图分别见图1-2)。参照美国医学遗传学和基因组学学会(ACMG)相关指南显示[5],M1和M2均为罕见致病变异,在gnomAD数据库中,M1在普通东亚人群中的突变频率为0.05%,M2目前尚未见报道。功能研究发现,这两种变异在次要内含子的正确剪接中起重要作用。
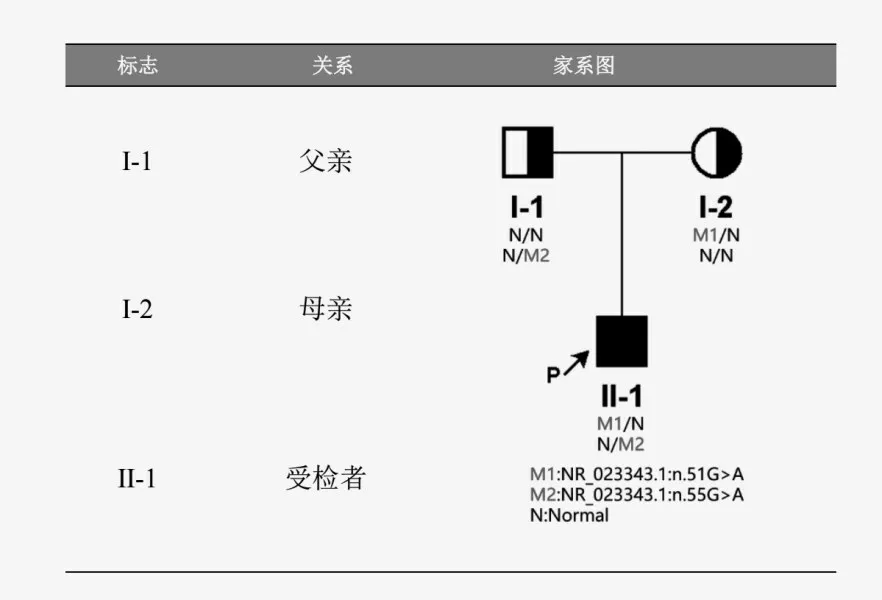
图1 患儿家系图
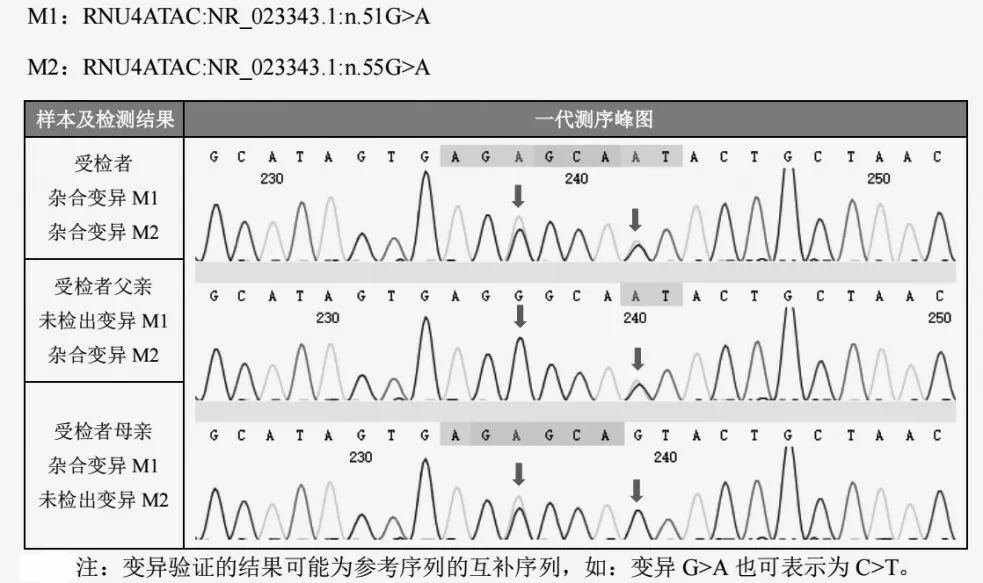
图2 一代测序峰图
2 讨论
小头畸形骨发育不良原始侏儒症Ⅰ型或Taybi-Linder综合征已被证明是由RNU4ATAC基因的双等位突变引起的,RNU4ATAC 基因位于染色体 2q14.2上CLASP1 基因的内含子2 中,被转录成一种非编码小核(sn)RNA,即U4atac snRNA。U4atac 是一种核糖核蛋白复合物,组成次要剪接体的重要部分,参与U12 型内含子的剪接[6]。目前大约在800 个基因中发现了U12 型内含子,这虽然只占人类基因组中所有内含子的0.5%。但多数含有 U12 型内含子的基因编码细胞离子通道或参与关键的细胞功能,例如 DNA 复制和修复、转录、RNA 加工和转运、翻译和细胞骨架组织[7],因此,次要内含子能否正确剪接在人类发育中起着重要作用。而功能分析表明,RNU4ATAC 中的突变将U12 依赖性剪接体活性降低了90%[8]。U4atac snRNP(snRNA 核糖核蛋白)通过将U6atac 加载到与U5snRNP 一致的含有U12 的剪接体前复合体上,U4atac 与U6atac 碱基配对的区域,即茎Ⅰ和茎Ⅱ(分别在U4atac的3” 和5” 处)由分子内茎环(5” 茎环)隔开,另一个茎环存在于U4atac 的3” 末端,其后是作为Sm 蛋白结合位点的序列,这对于snRNP 组装和输入细胞核很重要。茎Ⅱ、5” 茎环、茎I 和Sm 结合位点都富含高度保守的核苷酸,诱变实验证明了它们对正确的次要剪接体活性的重要性。后期研究发现,3” 茎环被认为有重要作用,据报道,3” 茎环的完全缺失消除了次要剪接体的体内剪接功能[9-10]。
RNU4ATAC 基因的双等位突变目前研究发现可以导致三种罕见的隐性发育疾病,即Taybi-Linder/MOPD1、Roifman 综合征(OMIM 616651)综合征和Lowry-Wood 综合征。RFMN 和LWS 均有与TALS重叠的特征(即小头畸形、生长迟缓、骨骼发育不良、智能障碍)。然而,在后两种疾病中,未观察到严重的脑结构异常和过早死亡,并且小头畸形和生长发育迟缓不那么明显[11]。另一方面,由于RFMN患者的父母首次咨询通常是因为他们的孩子反复感染,免疫缺陷问题已经在这种综合征中得到很好的证明,而TALS 和LWS 则不是这样。迄今为止,已经报道了12 种不同的RNU4ATAC 突变与TALS 相关。这些突变位于 U4atac/U6atac 复合物的U4atac 分子内5” 茎环(g. 30G>A,g. 40C>T, g. 46G>A, g. 50G>A, g.50G>C, g.51G>A、g.53C>G 和 g.55G>A)、茎I结构域(g. 66G>C)、分子内3” 茎环中(g. 111G>A,g. 16_100dup) 和附近的 U4atac Sm 蛋白结合位点(g.124G>A)[12],其中,g.51G>A 突变最常检测到,尽管没有明显的基因型—表型相关性,但与RNU4 ATAC 中其他描述的突变相比,双等位基因g.51G>A 突变与更短的寿命相关[13]。与其他原发性侏儒综合征相比,MOPD Ⅰ的预后较差。平均预期寿命约为8.5 个月,从2.5 个月到18 个月不等。早期死亡通常发生在1岁以内,主要由传染病引起。
在已报道的病例中,患儿主要表现为智力残疾和多种畸形,包括严重的小头畸形、皮质脑畸形(神经元移行缺陷)、胼胝体发育不全/发育不良、侏儒症和骨骼异常。报告的患者都存在胼胝体部分或完全缺失,其他的神经系统发育畸形包括额叶和颞叶发育不全、小脑蚓部发育不全、小脑半球相对保留和神经元迁移障碍(包括厚脑回、无脑回畸形、多小脑回和裂脑畸形)[13]。MOPD Ⅰ还可能表现出其他几个特征,如鱼鳞病、先天性心脏异常、小阴茎和隐睾、肾脏异常、听力障碍、先天性白内障和肝脾肿大[3]。既往Abdel-Salam 描述了5 例MOPD Ⅰ患者出现色素沉着异常:缺乏视网膜色素沉着、白发和皮肤白化病,以及患有脑出血和冻疮的血管病变患者,认为眼睛、头发和皮肤的异常色素沉着及血管病变也可能是该综合征的组成部分。但作者认为目前样本数量过少,结论真实性有待于进一步考证。
在本例报道中,受检者RNU4ATAC 基因中发现2个杂合变异,变异M1:n.51G>A 和变异 M2:n.55G>A,分别来自于母亲和父亲,出生时体质量:1 940 g,小于同胎龄儿出生体质量的第10 百分位,出生头围小(29 cm),现1 月27 天,头围31 cm,胸围36 cm,小头畸形,小颌畸形,耳位低平,皮肤干燥,毛发稀疏,双眼突出,孕检NT: 3.3 mm,唐筛18-三体临界风险,羊水穿刺提示样本的染色体在检测范围内未见异常,四维彩超提示胎儿颅骨内结构异常(BLAKE 囊肿待排?胼胝体缺失?左侧颅内囊实性回声,鼻骨发育不良,SD : 4. 1-4. 5 )。入院后查辅检头颅CT 检查示脑实质体积缩小,脑沟裂增深、增宽,双侧脑室分离,幕上、下脑室系统扩张,中线结构尚居中,为MOPDⅠ典型头颅畸形,家属因经济原因拒绝完善头颅MRI、脊柱X 线、骨盆正位X 线、双下肢X线及眼底检查,未能提供进一步病情评估,但随访未再出现病情加重现象。
MOPDⅠ迄今尚无特效治疗,本例报道丰富了其临床表型及基因谱。因RNU4ATAC 基因不是蛋白质编码基因,因此多数外显子组产品未能捕获到。未来随着全基因组测序过渡到临床领域,我们可能会看到这些诊断的逐渐增加,通过对RNU4 ATAC 基因突变患者的大量样本进行表型分析,不仅可更好地了解这种疾病的表型变异性,还可为建立这种罕见疾病的基因型—表型相关性提供进一步信息,有助于为患病家庭提供准确的复发风险指导。
猜你喜欢
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01 08:10:30
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
生物工程学报(2019年6期)2019-07-10 08:38:38
生物学通报(2019年1期)2019-02-15 16:33:43
生物学通报(2018年12期)2018-10-10 06:52:36
现代园艺(2017年21期)2018-01-03 06:41:32
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
百科知识(2015年18期)2015-09-10 07:22:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
